Abstract
The efficiency of metazoan origins of DNA replication is known to be enhanced by histone acetylation near origins. Although this correlates with increased MCM recruitment, the mechanism by which such acetylation regulates MCM loading is unknown. We show here that Cdt1 induces large scale chromatin decondensation that is required for MCM recruitment. This process occurs in G1, is suppressed by Geminin, and requires HBO1 HAT activity and histone H4 modifications. HDAC11, which binds Cdt1 and replication origins during S-phase, potently inhibits Cdt1-induced chromatin unfolding and re-replication, suppresses MCM loading, and binds Cdt1 more efficiently in the presence of Geminin. We also demonstrate that chromatin at endogenous origins is more accessible in G1 relative to S-phase. These results provide evidence that histone acetylation promotes MCM loading via enhanced chromatin accessibility. This process is regulated positively by Cdt1 and HBO1 in G1 and repressed by Geminin-HDAC11 association with Cdt1 in S-phase, and represents a novel form of replication licensing control.
Introduction
The initiation of DNA replication is regulated by a multi-subunit complex called the pre-Replication Complex (preRC) that assembles in a stepwise manner at chromosomal origins.Citation1 PreRCs are composed of the Origin Recognition Complex (ORC), which recruits two proteins, Cdc6 and Cdt1, both of which are required to load the hexameric Mini-Chromosome Maintenance (MCM) helicase.Citation2–Citation4 MCM loading occurs once during the cell cycle, and the events surrounding MCM loading are known collectively as replication licensing.Citation5 Whereas Cdc6 has been proposed to function as an MCM clamp loader,Citation6 the mechanisms by which Cdt1 promotes MCM loading are less clear. Cdt1 is known to be positively and negatively regulated by a small protein called Geminin.Citation7,Citation8 Although the binding of Geminin to Cdt1 is necessary for Geminin to affect Cdt1 function,Citation9 the molecular function of Cdt1 that Geminin regulates is also unclear.
DNA is packaged into nucleosomes that form higher-order chromatin structures. This organizes the genome but generates a physical barrier to accessing the DNA substrate for processes such as transcription and replication. Much of our understanding of how chromatin is accessed and manipulated is derived from transcriptional studies. The transcription apparatus modifies histones at promoters and within transcribed regions to create chromatin access. One such modification, acetylation, is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs).Citation10,Citation11 Acetylation of nucleosomes is generally associated with an open, accessible chromatin state that promotes transcription, while deacetylation counters this and produces a more closed, inaccessible state that reduces promoter usage and transcription.
Little is known about how the DNA replication machinery modulates chromatin structure to facilitate preRC assembly in eukaryotic cells. Studies using yeast and Drosophila have demonstrated that acetylation influences initiation timing and origin activity.Citation12,Citation13 Similarly, firing of the β-globin origin is temporally controlled by histone acetylation in mammalian cells.Citation14 Furthermore, the histone acetyltransferase HBO1 binds to mammalian origins through a physical interaction with Cdt1 and acetylates histone H4 tails at origin regions during G1, which is required for MCM recruitment.Citation15,Citation16 Although it is possible that this HBO1-induced acetylation controls MCM loading via chromatin structural changes and increased accessibility, the validity of this remains to be shown.
We show here that the mechanism whereby Cdt1 and HBO1 promote MCM loading in vivo involves the stimulation of large-scale chromatin decondensation to allow access to the underlying DNA substrate. We further show that the histone deacetylase HDAC11, whose physiological role in cells is poorly understood, counters this process and inhibits Cdt1-induced chromatin decondensation, MCM loading and re-replication. Intriguingly, Geminin enhances the binding of HDAC11 to Cdt1 and inhibits Cdt1-induced chromatin decondensation. These results provide evidence for a novel chromatin accessibility role for Geminin, Cdt1, HBO1 and HDAC11 in regulating replication licensing.
Results
HDAC11 associates with replication origins, inhibits Cdt1-induced re-replication and suppresses MCM loading.
HBO1 interacts with Cdt1 at origins specifically during G1 and acetylates H4 tails, which is required for MCM loading.Citation15,Citation16 The acetylation diminishes during S phase when MCM loading is normally prevented,Citation16 suggesting that a histone deacetylase may be involved in negatively regulating MCM loading. HDAC11 interacts directly with Cdt1 in S phaseCitation17 and can deacetylate H4 tailsCitation18,Citation19 but is poorly understood in terms of its physiological function in cells. As such, we asked if HDAC11 could bind to origins in S phase and negatively influence MCM loading and DNA replication. Chromatin immunoprecipitation (ChIP) analyses performed on two origins previously studied for HBO1 interactionsCitation15 demonstrated that HDAC11 becomes bound to MCM4 and Lamin B2 origins in S phase but not in G1, whereas nearby chromosomal regions show a small, but non-significant increase in HDAC11 (). Therefore, HDAC11 interacts with Cdt1 and associates with chromosomal origins with the opposite kinetics of HBO1 (i.e., during S phase), providing an explanation for why, in addition to reduced HBO1 activity, the H4 acetylation diminishes during S phase.Citation16
Co-expression of HBO1 enhances Cdt1-induced re-replication. Given that HDAC11 associates with Cdt1 and origins in S phase, we reasoned that HDAC11 might act in an opposite manner to HBO1 and suppress Cdt1-induced re-replication. Adenoviruses were used to overexpress Cdt1Citation20 at three different levels, which produced a dose-dependent increase in the percentage of cells with >4 N DNA content (). Co-expression of HDAC11 with Cdt1 caused a significant reduction in the number of re-replicating cells (, top parts). Interestingly, expressing more Cdt1 diminishes the inhibitory effects of HDAC11 (, bottom parts). This indicates that the suppression of DNA replication by HDAC11 is derived from a stoichiometric relationship that exists between the amount of Cdt1 and HDAC11 that is co-expressed. Furthermore, these results suggest that the inhibitory effect of HDAC11 is not due to an unrelated block to cell cycle progression into S phase. Overexpression of HDAC11 alone suppresses the loading of Mcm2 on chromatin () but has no effect on the total levels of Mcm2 or Cdt1. These results demonstrate that HDAC11 localizes to chromosomal origins in S phase and inhibits the ability of Cdt1 to promote DNA replication and MCM loading. As such, and given the positive role of HBO1 in these processes during G1,Citation15,Citation21 HDAC11 temporally opposes the function of HBO1 in regulating replication licensing via Cdt1 interactions in S phase.
Geminin facilitates the binding of HDAC 11 to Cdt1.
The ability of HDAC11 to bind to Cdt1, negatively influence MCM loading and suppress DNA replication is similar to the effects of Geminin.Citation9,Citation22 This suggested that a relationship might exist between Geminin and HDAC11 in regulating Cdt1 function. We obtained an anti-HDAC11 antibody that recognizes two isoforms of HDAC11, indicated here as bands A and B. Using synchronized cell lysates separated into soluble and chromatin bound fractions, we observed that Geminin and the faster migrating band B of HDAC11 became chromatin bound with similar kinetics specifically during S phase, both of which parallel PCNA binding kinetics (). The slower migrating band A of HDAC11 increases only modestly on chromatin during S phase. In contrast, HBO1 associates with chromatin earlier in G1 and peaks during the time of MCM loading (6–12 hrs), consistent with the positive role HBO1 enzymatic activity plays in promoting licensing during G1.Citation15,Citation16,Citation21 Cdt1 is chromatinbound throughout G1 and S phase, but a slower migrating form (asterisk) becomes visible that overlaps MCM loading kinetics (). The slower migrating Cdt1 is likely to be a ubiquitinylated form of Cdt1 that is known to be degraded.Citation23 Consistent with this, the slower migrating Cdt1 diminishes after its initial appearance. The kinetics for HBO1 and HDAC11 are graphed in . These results are consistent with a model in which HBO1 promotes licensing in G1 and HDAC11 prevents re-licensing during S phase, in both cases through association with Cdt1.
Although Geminin negatively influences the acetyltransferase activity of HBO1, it does not affect the physical interaction of HBO1 with Cdt1.Citation15 Given the similar chromatin binding kinetics between Geminin and HDAC11, we next determined whether Geminin influenced the interaction of HDAC11 with Cdt1. Geminin and HDAC11 can independently bind Cdt1 in vivo and in vitro,Citation9,Citation17 indicating that neither protein requires the other to directly bind Cdt1. HDAC11, Geminin and Cdt1 were transiently expressed in several combinations, and immunoprecipitations (IP) were performed against HDAC11 or Cdt1, followed by immunoblotting (IB) for the presence of the other expressed proteins in the IP complexes. Without Geminin, HDAC11 and Cdt1 interact to a small degree when either protein is pulled down in the IP step (, lane 5, rows A and C). Similarly, Geminin can bind Cdt1 in the absence of HDAC11 (, lane 6, row D). However, when all three proteins are co-expressed, there is a noticeable increase in the amount of HDAC11 that interacts with Cdt1 when either Cdt1 or HDAC11 is pulled down in the IP step ( and compare lanes 5 and 8 on rows A and C). The amount of Geminin that interacts with Cdt1 is not influenced by HDAC11, indicating that the converse is not true ( and compare lanes 6 and 8, row D). These results demonstrate that Geminin increases the efficiency of the HDAC11-Cdt1 interaction.
We next asked if Geminin, HDAC11 and Cdt1 could form a trimeric complex in cells or reside together in a larger protein complex. Cdt1, Geminin and HDAC11 were co-expressed and complexes containing Flag-HDAC11 were purified and separated by a size-exclusion column. All three proteins co-elute in a ∼700 kDa size range (fractions 15 and 16). Such an elution profile could be due to two separate but similarly-sized large complexes in which HDAC11 is present with Geminin in one and with Cdt1 in the other. However, this is highly unlikely given that Geminin and Cdt1 interact efficiently in cells on their own.Citation9,Citation22 Therefore, these results indicate that all three proteins reside together in one complex (that contains other unknown proteins), which is consistent with the fact that HDAC11 and Geminin both associate with Cdt1 in vivo during S phase under physiologic conditions.Citation17,Citation22 Since Geminin and HDAC11 do not reduce the efficiency with which either protein can bind Cdt1 (), Geminin and HDAC11 do not compete for binding to Cdt1 and can interact with Cdt1 simultaneously. These results suggest that one function of Geminin in negatively regulating DNA replication may derive from an inherent ability of Geminin to facilitate HDAC11 binding to Cdt1, leading to decreased MCM loading.
Cdt1 targeting induces large-scale chromatin decondensation.
Cdt1 recruits two histone modifying enzymes, HBO1 and HDAC11, that regulate MCM loading and DNA replication in an opposing manner. The timing of the association of these enzymes with replication origins coincides with the presence or absence, respectively, of acetylated histone H4.Citation16 Although the H4 acetylation is known to be required for MCM recruitment,Citation16 the mechanism by which it facilitates this is unknown. We hypothesized that the ability of Cdt1 to differentially recruit these enzymes produces higher-order chromatin structural changes that facilitate or inhibit MCM recruitment via altered chromatin accessibility. Currently, there is no technological means to assess changes to higher-order chromatin structure at chromosomal origins. However, to test this concept, we employed an innovative chromatin remodeling system that assesses the ability of proteins to generate changes to higher-order chromatin structure.Citation24,Citation25 This system utilizes a CHO-derived cell line (A03_1) that contains a 90 Mb homogeneous staining region (HSR) that was engineered through stable insertion and amplification of a Lac-operator(LacO)/DHFR vector (final HSR contains ∼1,600 such vectors). The presence of LacO sites throughout the HSR allows for microscopic visualization of chromatin structural changes that occur following targeting of LacI-fused proteins of interest. In its normal unperturbed state, the HSR adopts a condensed dot-like structure that is heterochromatic in nature.Citation26 However, targeting proteins that recruit chromatin remodeling enzymes elicits dramatic changes in the HSR structure resulting in clearly observable, highly decondensed HSRs occupying large portions of the nucleus.Citation24,Citation25 This system provides insight into regulation of higher-order chromatin dynamics that cannot be analyzed by any other current experimental means.
The mechanisms underlying chromatin remodeling in this system derive from specific, physiologically relevant events involved in altering chromatin structure by targeted proteins. Several transcription factors, including p53, E2F1, BRCA1, VP16 and ER, promote decondensation via histone acetylation, H2AX phosphorylation and recruitment of chromatin-modifying enzymes.Citation24,Citation27,Citation28 The replication protein Cdc45 promotes decondensation via Cdk2 recruitment and H1 phosphorylation.Citation25 In contrast, some proteins promote condensation,Citation29 while others produce no changes to the HSR structure (remains condensed).
To determine whether Cdt1 can promote large-scale decondensation of the HSR, Cdt1 was fused to LacI and transfected into A03_1 cells. As controls, LacI-VP16, LacI-Cdc6 or the LacI DNA binding domain (DBD) alone were also expressed. shows that LacI-VP16 promotes large-scale decondensation while LacI-Cdc6 and LacI-DBD do not, consistent with previous findings.Citation24,Citation25 Targeting Cdt1 to the HSRs produces a dramatic decondensation of the chromatin (). All proteins express similarly (), and the fact that LacI alone expresses significantly higher indicates that targeting proteins do not themselves elicit changes to the HSR due to crowding or related effects. As described previously,Citation25 we assigned “open” versus “closed” status to the visual appearance of the HSRs using objective criteria. Open structures clearly display large, decondensed HSRs that occupy more than 10% of the nuclear area. Closed HSRs are obvious condensed structures that failed to unfold and typically cover less than 5% of the nuclear area. In all analyses, some HSRs are visible that are dot-like in appearance, but somewhat larger in size (∼5–10% of nuclear area). We refer to the latter as Indeterminate since classifying such HSRs is highly subjective. Using these objective criteria, ∼2/3 of LacI-Cdt1-targeted HSRs become decondensed, similar to that for VP16 (). In addition to being enriched at the HSRs due to LacI targeting, the LacI-Cdt1 protein is also localized throughout the nucleus and not in the cytoplasm (), demonstrating that the localization of ectopic Cdt1 is regulated by physiologic mechanisms. We conclude from these results that targeting Cdt1, but not Cdc6, to chromosomal regions in vivo produces a clearly observable and robust decondensation of higher-order chromatin structure.
Cdt1-induced chromatin unfolding occurs during G1.
We reasoned that if chromatin unfolding induced by Cdt1 were physiologically involved in creating chromatin access for loading MCMs, then such unfolding should occur during G1. We determined the cell cycle phase at the time when decondensation occurred after LacI-Cdt1 targeting. To indicate S-phase cells, BrdU staining was used, and cells that were in G2 and/or M phases were identified by anti-H1-phospho (H1-P) staining since H1-P levels are highest at these times.Citation25,Citation30 LacI-Cdt1-induced open HSRs were found almost exclusively in transfected cells that neither displayed BrdU nor H1-P staining (). These results indicate that the cells are primarily in G1 (but early S-phase is also possible) at the same time that the transient LacICdt1 protein is expressed and open HSRs are being generated. Interestingly, closed HSRs correlated in the opposite manner (i.e., with S-, G2- or M-phase cells). Thus, chromatin unfolding by Cdt1 occurs in G1, when Cdt1 is known to function in MCM loading.
Geminin efficiently and specifically suppresses Cdt1-induced chromatin unfolding.
Since Geminin is a physiologic inhibitor of Cdt1 at high Geminin:Cdt1 ratios,Citation8 we asked if the decondensation by Cdt1 were Geminin sensitive. Chromatin decondensation assays were performed using a 1:1 ratio of Geminin:Cdt1 vectors or a higher 5:1 ratio. Relative protein expression is shown in . Compared to LacI-Cdt1 + pcDNA3 control, 1:1 ratios of Geminin:Cdt1 did not alter the amount of decondensation produced by Cdt1 (). However, co-expression of Geminin at a 5:1 ratio significantly suppressed the ability of Cdt1 to decondense chromatin ( and ). Under these conditions, we noticed the appearance of a number of very small but slightly decondensed HSRs, which we define as “small-open” (, left part). We considered these “small-open” HSRs as closed since they have clearly not succeeded in becoming the large, decondensed HSRs that are normally seen with Cdt1 expressed alone (compare and H). Chromatin unfolding induced by Cdc45 or VP16 was not sensitive to inhibition by Geminin (), indicating that the inhibitory effect of Geminin toward Cdt1 is specific and is not due to global effects that suppress chromatin remodeling mechanisms. These results demonstrate a novel effect of Geminin in modulating chromatin accessibility through its interaction with Cdt1.
Chromatin unfolding by Cdt1 is required for cell proliferation and efficient DNA re-replication.
We next determined the region within Cdt1 that is required for promoting chromatin unfolding and then tested for biological effects of loss of this domain. Carboxy-terminal truncations of Cdt1 were generated and tested for chromatin unfolding ability and it was found that a region in the middle of Cdt1 is required for chromatin decondensation. A deletion mutant of Cdt1 was made that lacked specifically this region () and was deficient for chromatin unfolding (). Stable expression of Cdt1-(Δ201-355) was found to significantly inhibit the ability of cells to proliferate relative to wt-Cdt1 (). Intriguingly, a previous report analyzing Cdt1 mutant alleles found that Cdt1 lacking this region is 25–60% less efficient at promoting re-replication versus multiple Cdt1 alleles that contain this region.Citation31 In agreement with this prior study, Cdt1-(Δ201-355) is 25–50% reduced in re-replication ability versus wt-Cdt1 (). The reason Cdt1 rereplication is not completely diminished is because all alleles containing the aminoterminus of Cdt1 will induce re-replication due to dilution of Cdt1 degradation components, allowing endogenous Cdt1 to induce re-replication in addition to the exogenous protein being tested.Citation31 As such, we conclude from these experiments that the chromatin unfolding function of Cdt1 is required for sustained cell cycle progression due at least in part to a necessity for this region to promote efficient DNA replication.
Chromatin decondensation by Cdt1 stimulates MCM recruitment.
We next asked if chromatin decondensation by Cdt1 stimulated the recruitment of endogenous MCMs. Chromatin unfolding assays were performed in which LacI-Cdt1 was expressed, followed by co-staining against LacI (to identify open or closed HSRs) and Mcm4 or Mcm7. and B show that endogenous Mcm4 and Mcm7 both become noticeably enriched at Cdt1-decondensed HSRs. In contrast, HSRs decondensed by BRCA1 or VP16 did not enhance Mcm7 recruitment ( and D). We also found that PCNA became enriched at Cdt1-decondensed HSRs (), but the effect was not dramatic and only occurred in a small percentage of such samples (<10%, data not shown).
Relative to BRCA1 and VP16, where MCM co-localization was far less frequent and not dependent on chromatin decondensation, ∼1/3 of Cdt1-decondensed HSRs displayed enriched MCM recruitment (). Only a small number (5%) of Cdt1-bound HSRs that failed to open recruited MCMs. This result was obtained in more than six separate experiments (data not shown, but see below). This consistent observation probably derives from our necessary use of asynchronous populations for these analyses. The machinery involved in MCM loading is only available during a certain period of time in the cell cycle, and in cells released from quiescence, MCM loading occurs in the latter ∼1/3 of G1.Citation32 Cdt1-induced decondensation occurs in G1 () and MCM recruitment is seen in only ∼1/3 of these, which correlates with such a prediction.
A simple explanation for why MCMs are enriched at the HSRs upon Cdt1 targeting could derive from the fact that Cdt1 can bind to MCMs.Citation4,Citation31,Citation33 However, Cdt1-bound HSRs that fail to open are not efficiently enriched with MCMs (), indicating that the presence of Cdt1 alone at these chromosomal sites is not sufficient for MCM recruitment. Since a significant number of Cdt1-decondensed HSRs are not enriched for MCMs, the recruitment of MCMs does not itself produce a crowding effect that causes the unfolding. We conclude from these results that Cdt1-induced decondensation is a prerequisite for stimulating MCM recruitment.
HBO1 and HDAC11 regulate Cdt1-induced chromatin unfolding.
Given that HBO1 and HDAC11 are known histone/chromatin modifiers,Citation18,Citation34 we asked whether these factors could modulate Cdt1-induced chromatin decondensation. We verified that HDAC11 and Cdt1 interact in vivo in reciprocal co-IP experiments (). Similarly, HBO1 and Cdt1 interact in vivo (). LacI-Cdt1 was co-expressed with HDAC1, HDAC11, HBO1-wt or HBO1G485A (catalytically-inactive), and the decondensation potential of Cdt1 was determined for each condition. Similar amounts of LacI-Cdt1 expression were achieved, but more LacI-Cdt1 was expressed with HBO1G485A (). Similar expression of HDAC1 and HDAC11 was achieved, while HBO1G485A expressed slightly less well compared to HBO1-wt. HDAC1 and HBO1-wt do not alter the ability of Cdt1 to induce chromatin unfolding (). However, HDAC11 dramatically suppresses the ability of Cdt1 to cause decondensation, producing a concomitant increase in closed HSRs. Despite being expressed at lower levels relative to HBO1-wt and in the presence of higher amounts of LacI-Cdt1, HBO1G485A also suppresses Cdt1-induced decondensation. In comparison, HDAC1, HDAC11, HBO1-wt and HBO1G485A do not affect VP16-induced decondensation (). We conclude from these results that HBO1 normally performs a positive role specifically in Cdt1-induced chromatin unfolding, while HDAC11 is a strong and specific inhibitor of the decondensation by Cdt1. Furthermore, these results indicate that the effects of HBO1 and HDAC11 on chromatin remodeling by Cdt1 are not due to global cellular changes that affect chromatin remodeling in general.
HBO1 and HDAC11 influence MCM recruitment to Cdt1-targeted HSRs.
We next asked if HBO1 or HDAC11 influenced the level of MCM recruitment to Cdt1-targeted HSRs. Chromatin remodeling assays were performed as above, but costained and quantified for enrichment of endogenous Mcm4 or Mcm7. HDAC1 and HBO1-wt again had no effect on the ability of Cdt1 to cause chromatin decondensation (data not shown), nor did either protein significantly alter the amount of Mcm4 or Mcm7 that was enriched overall ( and ). In both cases, MCM enrichment was primarily associated with HSRs that had undergone a decondensation event (). In contrast, HDAC11 and HBO1G485A, again, inhibited the ability of Cdt1 to cause decondensation (producing closed HSRs, data not shown), and this was associated with a significant reduction in total MCM enrichment ( and ). For both HDAC11 and HBO1G485A, any enrichment of MCMs was almost exclusively associated with the small percentage of HSRs that had unfolded under these conditions (data not shown). We conclude from these results that HBO1 is normally required for efficient chromatin unfolding and MCM recruitment by Cdt1, while HDAC11 is a potent and specific inhibitor of the ability of Cdt1 to cause decondensation and MCM recruitment.
Chromatin decondensation and MCM recruitment by Cdt1 involve histone H4 acetylation.
Recruitment of MCMs to chromosomal origins depends on HBO1 acetyltransferase activity toward histone H4.Citation16 We reasoned that histone H4 modifications played a role in the Cdt1-induced decondensation and MCM recruitment to the HSRs due to the involvement of HBO1 and HDAC11 in this process. Although we predicted that H4 acetylation on residues K5, K8 or K12 should be increased at the decondensed HSRs following Cdt1 targeting, we observed no stable association of such modifications with the unfolded HSRs (data not shown). H4 acetylation is known to be a transient event at origins,Citation16 which likely explains our inability to observe stable H4 modifications. However, to show that H4 acetylation does play a functional role in the decondensation process, we took advantage of the ability of the Set8 histone methylase H4 binding domain (HBD) to interact with H4 tails and block their acetylation.Citation16,Citation35
Set8-HBD, HDAC1 or GST was co-expressed with LacI-Cdt1, and the ability of Cdt1 to promote chromatin decondensation and MCM recruitment was determined. As described above, HDAC1 does not affect the ability of Cdt1 to unfold chromatin and promote MCM recruitment. However, although Set8-HBD expresses less efficiently than HDAC1 (), co-expression of Set8-HBD significantly reduces the ability of Cdt1 to promote decondensation relative to HDAC1 ( and F and ). Co-expression of GST similarly has no effect on Cdt1-induced chromatin unfolding ( and ) and MCM recruitment ( and ). These results strongly suggest that, at least in a transient manner, acetylation of H4 tails is necessary for Cdt1 to induce chromatin unfolding and stimulate MCM recruitment in vivo. These findings are consistent with prior studies showing that HBO1 catalytic activity and the resultant H4 acetylation at origins are required for MCM recruitment by Cdt1,Citation16 but now we provide mechanistic evidence that such H4 acetylation promotes chromatin accessibility and fluidity that facilitates the loading of the MCM complex. Intriguingly, the amount of suppression elicited by Set8-HBD is very similar to that caused by HBO1G485A (), as expected if H4 acetylation by HBO1 plays a functional role in Cdt1-induced chromatin unfolding. However, neither Set8-HBD nor HBO1G485A are as potent as HDAC11 at suppressing Cdt1-induced unfolding, suggesting that additional modifications, perhaps on other histone subunits, are likely involved in this process.
Chromatin at endogenous origins of DNA replication is more accessible during G1 versus S phase.
Our results suggest that chromatin at origins of DNA replication will be more accessible in G1, when MCMs are loading, due to Cdt1 and HBO1 activities, but less accessible during S phase due to HDAC11 recruitment by Cdt1. Intriguingly, at least three reports in the literature have shown this situation to be true at higher eukaryotic origins. The chromatin at the GAS41 origin in chicken cells and the β-globin origin in human cells displays increased DNase I hypersensitivity during G1, but becomes less accessible to nuclease digestion in S phase.Citation36,Citation37 Similarly, chromatin at the ori-β and ori-γ origins in CHO cells is more accessible and sensitive to micrococcal nuclease in G1 versus S phase.Citation38 We determined whether the same were true at the MCM4 and Lamin B2 origins in human cells. HaCaT cells were synchronized and released into the cell cycle and intact chromatin was isolated in late-G1 and S phase and subjected to controlled DNase I digestion followed by qPCR analysis (). Less accessible chromatin at these origins reduces DNase I digestion, resulting in more substrate available for qPCR. Relative to late-G1, the MCM4 and Lamin B2 origins are both less accessible to DNase I in S phase, as indicated by the increased qPCR substrate availability from these time points. Thus, six higher eukaryotic endogenous replication origins analyzed by different methods (indirect end labeling or qPCR) display increased chromatin accessibility in G1 but less accessibility during S phase. Our results now provide a molecular explanation for this differential chromatin accessibility at replication origins that involves Cdt1-modulated control over higher-order chromatin structure via temporal recruitment of HBO1 and HDAC11.
Discussion
We present evidence for a novel form of replication licensing control that involves the ability of Cdt1 to modulate chromatin accessibility through the temporal recruitment of HBO1 and HDAC11 (modeled in ). In G1, Cdt1 (via ORC interaction) recruits HBO1 to replication origins, resulting in acetylation of H4 within the origin regions.Citation15,Citation16 We show here that at least one effect of this acetylation is an increase in chromatin accessibility that is required for MCM recruitment. HBO1 catalytic activity is required for MCM loading at origins,Citation16 and HBO1 stimulates Cdt1-dependent re-replication.Citation15 Upon entering S phase, de novo MCM recruitment is blocked, and we show here that HDAC11 contributes to this process. HDAC11 interacts with Cdt1 and localizes to replication origins specifically in S phase, and HDAC11 is capable of catalyzing the removal of acetylation from H4.Citation18 Consistent with this, H4 acetylation decreases at replication origins during S phase.Citation16 HDAC11 potently inhibits the ability of Cdt1 to cause chromatin decondensation, suppresses the recruitment of MCMs and blocks Cdt1-induced re-replication. As such, HDAC11 directly opposes the functions of Cdt1 and HBO1 in promoting replication licensing, thereby producing a “yin-yang” relationship between HBO1 and HDAC11. The mechanism underlying this relationship derives from the ability of HBO1 to promote chromatin decondensation for MCM loading in G1, followed by the recruitment of HDAC11 in S phase, which produces chromatin inaccessibility and prevents MCM loading. Such a model is supported by temporal changes in chromatin accessibility at endogenous origins in higher eukaryotic cells, shown here and by others,Citation36–Citation38 where origins are more accessible in G1 and transition to less accessible chromatin organization in S phase.
Geminin is a physiologic inhibitor of Cdt1 during S phase.Citation22 While the binding of Geminin to Cdt1 reduces the ability of Cdt1 to interact with the MCM complex,Citation33,Citation39 Geminin has been found to also influence the function of HBO1 in association with Cdt1. Geminin does not alter the interaction of HBO1 with Cdt1Citation15 but instead inhibits the acetyltransferase activity of HBO1.Citation16 We present evidence here that another mechanism whereby Geminin modulates Cdt1 function is through enhanced HDAC11 recruitment to Cdt1 (modeled in ). Thus, Geminin indirectly suppresses H4 acetylation at origins by inhibiting HBO1 acetyltransferase activity and by promoting the recruitment of HDAC11. As a result, Geminin produces decreased chromatin accessibility that blocks MCM loading, which is supported by our observation that Geminin potently and specifically suppresses the chromatin decondensation induced by Cdt1. Currently, we do not know how Geminin modulates HBO1 HAT activity or HDAC11 association with Cdt1. One possibility is that Geminin directly influences HBO1 activity and interactions of HDAC11 with Cdt1, although Geminin does not compete with either protein for Cdt1 binding. However, an alternative explanation may derive from Geminin-regulated recruitment of unknown factors that themselves control these events. Clearly, further investigation is required to answer these questions.
There is currently no technological means to observe largescale chromatin structural changes at specific single genomic loci in mammalian cells (i.e., origins). Although we have utilized an innovative, but engineered, chromatin remodeling system to address this question, several lines of evidence indicate that the events observed using this system recapitulate those occurring at origins, but at a macroscopic level. Chromatin decondensation induced by Cdt1 occurs during G1 and is sensitive to Geminin in a highly specific manner. Cdt1-induced decondensation involves H4-Ac during the process of unfolding, is dependent on HBO1 function and is sensitive to HDAC11 inhibition. In both cases, these enzymes elicit their effects specifically for chromatin decondensation derived from Cdt1. MCM recruitment is clearly observed as a specific result of Cdt1-induced decondensation, and is inhibited by mutant HBO1 and HDAC11 in a specific manner. As described above, Cdt1, HBO1 and H4-Ac dependency for MCM recruitment is also true at origins specifically during G1. HDAC11 associates with origins during S phase (and not in G1), when H4-Ac decreases and origin access would be predicted to be blocked, and HDAC11 reduces Cdt1-induced re-replication potential and suppresses genomic MCM loading. Most importantly and highly consistent with our findings, endogenous DNA replication origins display temporal changes in chromatin organization that produce more open and accessible conditions during G1 but transition to a less accessible chromatin state in S phase (results herein and reviewed in ref. Citation36–Citation38). Collectively, such results provide a strong argument that our observations in vivo at this engineered locus represent physiologic events occurring during replication licensing that cannot be seen by any other current experimental approach. Importantly, these results indicate that chromatin accessibility is at least one mechanism whereby Cdt1, HBO1, HDAC11 and Geminin regulate replication origins via H4-Ac changes.
In yeast and flies, the HBO1 homologues GCN5 and Chameau, respectively, induce acetylation of histones globally and near origins, which promotes origin firing.Citation12,Citation13 In contrast, Rpd3, which is homologous to HDAC11, decreases acetylation and reduces origin activity.Citation12,Citation13 Similarly, the timing of activation of the β-globin origin in mammalian cells is influenced by its acetylation state. Whereas acetylation of the β-globin is associated with earlier firing, targeting HDAC2, which is related to HDAC11, renders the origin late-firing.Citation14 Our results provide a mechanistic explanation for these studies of replication origin control by histone acetylation in which the acetylation influences chromatin accessibility for MCM loading. Although we do not know biochemically how H4 acetylation promotes chromatin unfolding, at least two possibilities are likely. First, histone H4 acetylation has been shown to directly enhance chromatin accessibility via structural changes,Citation40 consistent with what we have observed herein. Second, histone acetylation may recruit bromodomain-containing proteins that facilitate the chromatin unfolding.Citation41 Neither of these mechanisms is mutually exclusive with the other, and it is possible both may contribute to chromatin structural changes at origins. Finally, it is likely that chromatin-modifying enzymes other than HBO1 and HDAC11 are involved in regulating replication origins. For example, SNF2H and WSTF have been shown to co-purify with Cdt1 and differentially bind to chromatin depending on histone tail modifications.Citation42,Citation43
Cdt1 is oncogenic and overexpressed, sometimes via amplification, in several human cancers, including lung and colon carcinomas, melanomas and some leukemias and lymphomas.Citation44–Citation46 The oncogenic nature of Cdt1 derives from its ability to promote MCM loading and re-replication, the result of which is an increase in genomic instability.Citation20,Citation46 Our results suggest that one molecular mechanism mediating Cdt1's ability to promote re-replication is the temporal recruitment of histone-modifying enzymes that alter chromatin structure and thereby modulate chromatin accessibility. Excessive levels of Cdt1 will inappropriately cause chromatin decondensation cycles at origins, allowing re-loading of MCMs within one cell cycle. The resultant reinitiation of DNA replication within S phase produces genomic instability, and, as such, provides a novel molecular explanation for how tumorigenesis can occur due to changes in chromatin accessibility at replication origins.
Materials and Methods
Cell culture and transfections.
Cells were maintained in MEM/10% Fetal Clone II (CHO, A03_1, HeLa) or DMEM/10% FBS (HaCaT, 293T). A03_1 also contained 0.3 µM methotrexate. Cells were synchronized by isoleucine deprivation (CHO) or serum deprivation (HaCaT) as described.Citation25,Citation47 Replicating DNA was labeled with 15 µM BrdU. Transfections were performed for 24 hrs with FuGene-6 (Roche). Adenovirus assays were performed as described.Citation20 Colony forming assays used pTK-Hygro co-transfected and hygromycin selection (400 µg/ml).
Antibodies.
Anti-LacI (Stratagene or Upstate); anti-BrdU (Roche); anti-H1P (provided by C. Mizzen, University of Illinois); anti-HBO1 (provided by M. Smith, University of Virginia); anti-Geminin, anti-Myc (S. Cruz Biotech); anti-HDAC11, anti-Flag, anti-actin (Sigma); anti-PCNA, anti-tubulin (Calbiochem); anti-HA (Covance); anti-Cdt1 (provided by H. Nishitani, Kyushu University, Japan); anti-Mcm2, anti-Mcm4 and anti-Mcm7 were generated by Covance or Aves Labs.Citation32,Citation47
Plasmids and cDNAs.
HsCdt1, HsGeminin and pEBG-GST were provided by A. Dutta (University of Virginia). HsCdt1, CgCdc45, CgCdc6, BRCA1 (6c-w mutant), HsHDAC1 and HsHDAC11 were expressed using pRcLac.Citation25 No NLS sequence was added to any LacI construct. LacI-VP16 was provided by A. Belmont (University of Illinois). HBO1-wt and HBO1.G485A were provided by M. Smith (University of Virginia). Geminin, Cdc6, HBO1-wt and HBO1-G485A were expressed from pcDNA3-HA and HBO1-wt. HDAC1 and HDAC11 were expressed from pcDNA3-Flag. Cdt1 was expressed using pcDNA3-6xMyc. Set8-HBD was generated by proofreading PCR and expressed using pcDNA3-HA-NLS.
Protein chemistry.
Immunoprecipitations (IP) were performed in TNN (50 mM Tris, pH 7.4, 250 mM NaCl, 0.1% Igepal CA-630 and phosphatase and protease inhibitors). Immune complexes were washed with lysis buffer 3X. For immunoblots, equal numbers of cells were lysed and boiled in loading dye [for total lysates (TCE)] or were separated into detergent-resistant (chromatin) or detergent-soluble fractions as described.Citation25,Citation48 PreRC subunits present in the chromatin fraction are sensitive to nuclease digestion.Citation48 TCE, soluble and/or chromatin samples representing equivalent cell numbers were analyzed by standard immunoblotting and ECL. For gel filtration, transfected cells were collected and lysed in buffer containing 0.2% NP-40. Lysates were purified over an anti-Flag column (Sigma) and eluted using a Flag peptide. Eluates were subjected to a size exclusion column (Superdex 200 HR 10/30 column, GE) using FPLC and fractions were collected and analyzed by immunoblotting.
Immunofluorescence and flow cytometry.
Cells were analyzed by IF and BrdU incorporation as described.Citation25 Photographs of cells were obtained with a Zeiss fluorescence microscope and images were merged using Adobe Photoshop. For flow analysis cells were washed, fixed with ethanol and stained with propidium iodide (PI) in Triton X-100 and RNAse.
ChIP assays and qPCR.
Synchronized HaCaT cells were fixed with 1% formaldehyde for 10 min at room temperature. Crosslinked chromatin was sonicated in 10 mM Tris-HCl (pH 8), 1 mM EDTA, 0.5 mM EGTA, 1% SDS (plus phosphatase and protease inhibitors) to an average length of ∼500 bp. Samples were adjusted to 5 mM Tris-HCl (pH 8), 30 mM NaCl, 0.2% Triton X-100, 0.2% SDS, 0.8% BSA, 0.4 mM EDTA, 0.1 mM EGTA and chromatin from 5 × 106 cells was used for IP with anti-HDAC11 or control IgG (4°C overnight). Immune complexes were precipitated with anti-rabbit agarose, washed and eluted in 10 mM Tris-HCl (pH 8), 1 mM EDTA, 1% SDS at 65°C. Crosslinks were reversed at 65°C overnight and samples were treated with Proteinase K for 3 hr at 50°C. Resulting DNA was purified using phenol/chloroform extractions and subjected to quantitative PCR (qPCR) in triplicate using a BioRad MyIQ detection system with TaqMan primers and probes against previously described origin sequences.Citation49,Citation50 Primers are available upon request. As previously described,Citation51 the enrichment of specific genomic DNA sequences was determined based on the threshold cycle (Ct) for each PCR product and analyzed according to the formula 2−[ΔCt(IP) − Ct(input)]-2−[ΔCt(control IgG) − Ct(input)]. Using this method, DNA relative to input and immunoprecipitated by anti-HDAC11 was normalized to DNA immunoprecipitated by control IgG. P values were obtained using the Student's two-tailed t-test.
DN ase I accessibility assays.
Chromatin was isolated in a buffer containing 10 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM CaCl2, 10 mM KCl, 300 mM sucrose and 0.1% Triton X-100 for 5 min on ice then washed and resuspended with the same buffer lacking detergent. One third of the chromatin from a 10 cm plate of cells was digested with DNase I (Promega) at 3 Units/100 µl for 10 min at RT. Another third was treated identically, but without DNase I (used for nomalization, untreated control). Reactions were stopped by addition of 10 mM EDTA/2 mM EGTA and incubated at 65°C for 10 min. DNA was lightly sonicated, then purified and analyzed using TaqMan-based qPCR as described for ChIP assays.
Figures and Tables
Figure 1 HDAC11 binds origins in S phase, inhibits Cdt1-induced re-replication and suppresses MCM loading. (A) Synchronized HaCaT cells (verified by BrdU incorporation, top) were subjected to anti-HDAC11 ChIP and qPCR analysis at the indicated times for interactions to origin and non-origin sequences. (B and C) Cdt1 alone or Cdt1 plus HDAC11 was expressed in HeLa cells for 48 h with adenoviruses and subjected to flow cytometric analysis. Relative amounts of each virus and the percentage of cells with >4 N DNA are indicated. (D) Mcm2 chromatin binding was assessed by IB in HeLa cells infected with HDAC11 or control (GFP) viruses for 24 h. (E) Synchronized CHO cells were separated into soluble and chromatin fractions at the indicated times and subjected to IB. BrdU verified synchrony (data not shown). (F) Graphical representation of IB results from (E). (G) Indicated proteins (top) were expressed in 293T cells and subjected to IP and IB analysis indicated on right. IP and IB analyses were performed with anti-tag antibodies. (H) Flag-HDAC11, HA-Geminin and Myc-Cdt1 were co-expressed in 293T cells followed by anti-Flag purification. Eluates were separated on a size-exclusion column and analyzed by IB. The data in (G and H) are representative of three independent experiments with similar results.
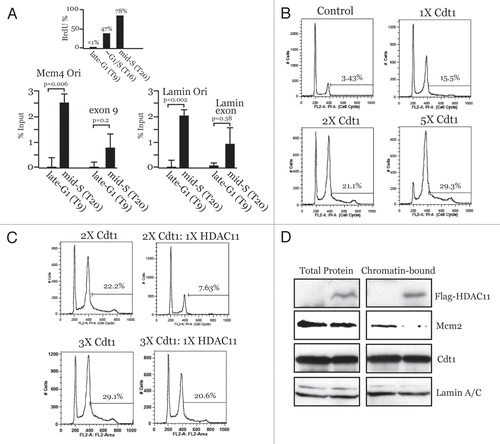

Figure 2 Cdt1 targeting induces Geminin-sensitive large-scale chromatin decondensation in G1. A03_1 cells were used in (A–J). (A) LacI-VP16, LacI alone or LacI-Cdc6 were transiently expressed, followed by IF with anti-LacI and Texas Red to detect open/decondensed (‘O’) or closed/condensed (‘C’) HSRs. Nuclei are stained with DAPI. (B) LacI-Cdt1 was expressed and analyzed by IF to detect chromatin decondensation. (C) Immunoblot of LacI-fusion protein expression for the results in . (D) Anti-LacI IF separated from DAPI showing LacI-Cdt1 present throughout the nucleus. (E) LacI-Cdt1 was expressed for 24 h, then pulsed with BrdU. Anti-BrdU and anti-H1-P staining was used to relate the index of BrdU-negative and H1-P-positive cells to the open or closed HSR status. (F) HA-Geminin was transfected at a 5:1 or 1:1 plasmid ratio with LacI-Cdt1 and relative protein expression verified by IB. (G and H) Examples of small-open, closed and large-open HSRs for the indicated conditions. (I) Diagram showing location of Cdt1 chromatin unfolding domain. (J) Chromatin unfolding ability of Cdt1-(Δ201-355) was tested as above. (K) Colony forming assays were performed in CHO cells to test the ability of wt-Cdt1 and Cdt1-(4201-355) to suppress colony growth. Stable selection for protein expression lasted 14 days, followed by Giemsa staining. (L) HeLa cells were used as in to determine the re-replication ability of Cdt1-(Δ201-355) versus wt-Cdt1 except 48-h transient transfections were used. Results from two experiments are shown compared to wt-Cdt1 (normalized to 100% rereplication ability).
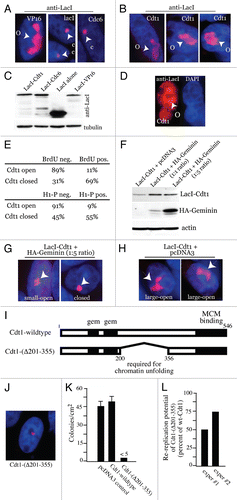
Figure 3 Cdt1-induced chromatin unfolding stimulates MCM recruitment. A03_1 cells were used in all parts. (A–E) Open HSRs following LacI-Cdt1, LacI-BRCA1 or LacI-VP16 expression were co-stained by IF with antibodies to LacI, Mcm4, Mcm7 or PCNA. Arrows indicate open HSRs and enrichment (or lack thereof) of endogenous MCMs or PCNA. (F) Quantification of endogenous Mcm7 recruitment to open or closed HSRs targeted by LacI-Cdt1, LacI-BRCA1 or LacI-VP16.
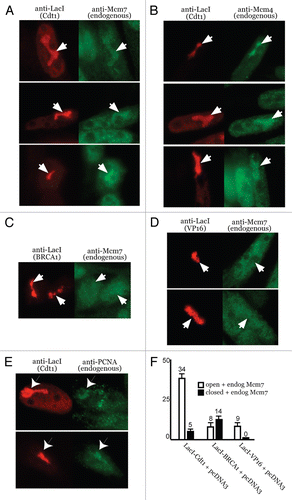
Figure 4 Cdt1-induced decondensation and MCM recruitment requires HBO1, is inhibited by HDAC11 and involves H4 acetylation. 293T cells were used in (A and B) and A03_1 cells were used in all others. (A and B) IP-western assays were performed using the indicated proteins and anti-tag antibodies. (C) Immunoblot of indicated proteins showing their relative protein expression for the results in and . (D) Examples of co-localizing Mcm7 (or lack thereof) in cells expressing indicated proteins. Samples were processed by IF with indicated antibodies as in . Open/decondensed (‘O’), closed/condensed (‘C’) HSRs. (E) Anti-Flag immunoblot showing relative transient expression of Flag-HDAC1 versus Flag-Set8-HBD. Parts are from the same immunoblot/exposure. (F) Examples of co-localizing Mcm4 (or lack thereof) in cells expressing indicated proteins. Quantitative results are presented in for (D and F).
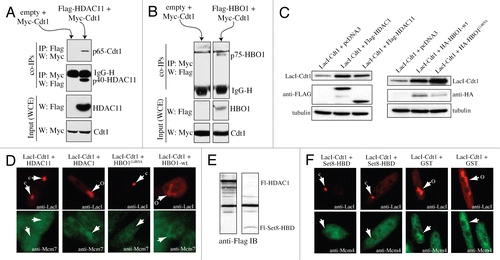
Figure 5 Chromatin at endogenous DNA replication origins is more accessible during G1 than in S phase. HaCaT cells were synchronized by serum deprivation and verified by BrdU incorporation as in . qPCR was performed on DNase I-treated chromatin samples from the indicated time points. Results were normalized against input chromatin from each time point that was not treated with DNase I to account for increases in DNA levels during S phase. Assays were performed in triplicate to generate error bars.
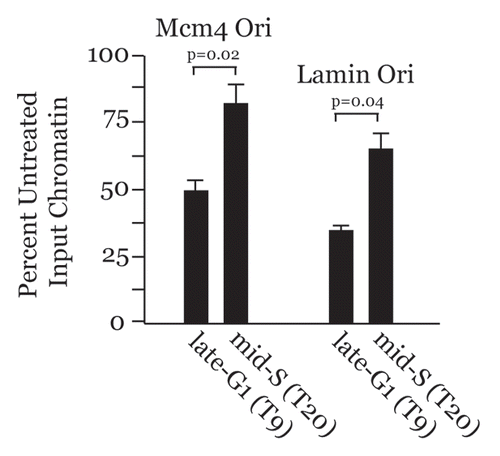
Figure 6 Model for replication licensing regulation via chromatin accessibility influenced by Cdt1, HBO1, HDAC11 and Geminin. See text for descriptions.
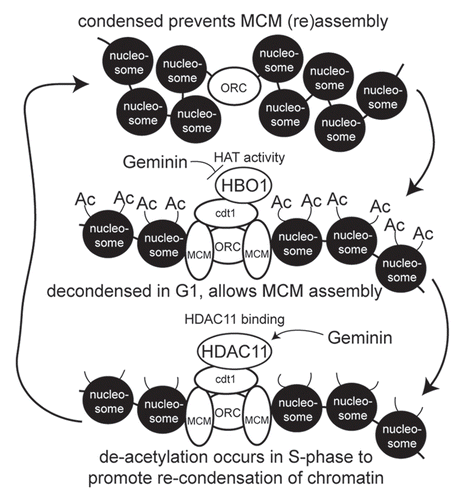
Table 1 Quantification of open and closed chromatin structures/HSRs
Table 2 Quantification of effects of HBO1, HDAC11 and Set8-HBD on chromatin unfolding
Table 3 Quantification of MCM colocalization with HSRs
Acknowledgements
We thank C. Mizzen, M. Smith and H. Nishitani for antibodies. We also thank A. Dutta, M. Smith and A. Belmont for cDNAs. P.G.W. was supported by a fellowship from the American Heart Association. M.G.A. was supported by the National Functional Genomics Center at the Moffitt Cancer Center, the Florida Department of Health and James and Esther King Foundation (Award 06-NIR-01), an NCI Lung SPORE (P50-CA119997) and a grant from the NCI (CA130865).
References
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem 2002; 71:333 - 374
- Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 1992; 357:128 - 134
- Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JFX. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature 1996; 379:180 - 182
- Tanaka S, Diffley JF. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat Cell Biol 2002; 4:198 - 207
- Blow JJ, Hodgson B. Replication licensing—defining the proliferative state?. Trends Cell Biol 2002; 12:72 - 78
- Perkins G, Diffley JFX. Nucleotide-dependent prereplicative complex assembly by Cdc6p, a homolog of eukaryotic and prokaryotic clamp-loaders. Molecular Cell 1998; 2:23 - 32
- Ballabeni A, Melixetian M, Zamponi R, Masiero L, Marinoni F, Helin K. Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. EMBO J 2004; 23:3122 - 3132
- Lutzmann M, Maiorano D, Mechali M. A Cdt1-geminin complex licenses chromatin for DNA replication and prevents rereplication during S phase in Xenopus. EMBO J 2006; 25:5764 - 5774
- Saxena S, Yuan P, Dhar SK, Senga T, Takeda D, Robinson H, et al. A dimerized coiled-coil domain and an adjoining part of geminin interact with two sites on Cdt1 for replication inhibition. Mol Cell 2004; 15:245 - 258
- Carrozza MJ, Utley RT, Workman JL, Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet 2003; 19:321 - 329
- Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007; 26:5310 - 5318
- Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. Histone acetylation regulates the time of replication origin firing. Mol Cell 2002; 10:1223 - 1233
- Aggarwal BD, Calvi BR. Chromatin regulates origin activity in Drosophila follicle cells. Nature 2004; 430:372 - 376
- Goren A, Tabib A, Hecht M, Cedar H. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev 2008; 22:1319 - 1324
- Miotto B, Struhl K. HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev 2008; 22:2633 - 2638
- Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell 2010; 37:57 - 66
- Glozak MA, Seto E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J Biol Chem 2009; 284:11446 - 11453
- Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 2002; 277:25748 - 25755
- Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol 2009; 10:92 - 100
- Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, et al. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell 2003; 11:997 - 1008
- Iizuka M, Matsui T, Takisawa H, Smith MM. Regulation of replication licensing by acetyltransferase Hbo1. Mol Cell Biol 2006; 26:1098 - 1108
- Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 2000; 290:2309 - 2312
- Arias EE, Walter JC. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev 2005; 19:114 - 126
- Tumbar T, Sudlow G, Belmont AS. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J Cell Biol 1999; 145:1341 - 1354
- Alexandrow MG, Hamlin JL. Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation. J Cell Biol 2005; 168:875 - 886
- Li G, Sudlow G, Belmont AS. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: Precise choreography of condensation/decondensation and nuclear positioning. J Cell Biol 1998; 140:975 - 989
- Ye Q, Hu YF, Zhong H, Nye AC, Belmont AS, Li R. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J Cell Biol 2001; 155:911 - 921
- Nye AC, Rajendran RR, Stenoien DL, Mancini MA, Katzenellenbogen BS, Belmont AS. Alteration of largescale chromatin structure by estrogen receptor. Mol Cell Biol 2002; 22:3437 - 3449
- Verschure PJ, van der Kraan I, de Leeuw W, van der Vlag J, Carpenter AE, Belmont AS, et al. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol Cell Biol 2005; 25:4552 - 4564
- Lu MJ, Dadd CA, Mizzen CA, Perry CA, McLachlan DR, Annunziato AT, et al. Generation and characterization of novel antibodies highly selective for phosphorylated linker histone H1 in Tetrahymena and HeLa cells. Chromosoma 1994; 103:111 - 121
- Teer JK, Dutta A. Human Cdt1 lacking the evolutionarily conserved region that interacts with MCM2-7 is capable of inducing re-replication. J Biol Chem 2008; 283:6817 - 6825
- Mukherjee P, Cao TV, Winter SL, Alexandrow MG. Mammalian MCM loading in late-G(1) coincides with Rb hyperphosphorylation and the transition to posttranscriptional control of progression into S-phase. PLoS ONE 2009; 4:e5462
- Yanagi K, Mizuno T, You Z, Hanaoka F. Mouse geminin inhibits not only Cdt1-MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J Biol Chem 2002; 277:40871 - 40880
- Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 2006; 21:51 - 64
- Yin Y, Yu VC, Zhu G, Chang DC. SET8 plays a role in controlling G1/S transition by blocking lysine acetylation in histone through binding to H4 N-terminal tail. Cell Cycle 2008; 7:1423 - 1432
- Djeliova V, Russev G, Anachkova B. DNase I sensitive site in the core region of the human beta-globin origin of replication. J Cell Biochem 2002; 87:279 - 283
- Zimmermann K, Holtz M, Phi-van L. The chromatin structure of the lysozyme GAS41 origin of DNA replication changes during the cell cycle. Biol Res 2007; 40:185 - 192
- Pemov A, Bavykin S, Hamlin JL. Attachment to the nuclear matrix mediates specific alterations in chromatin structure. Proc Natl Acad Sci USA 1998; 95:14757 - 14762
- Cook JG, Chasse DA, Nevins JR. The regulated association of Cdt1 with minichromosome maintenance proteins and Cdc6 in mammalian cells. J Biol Chem 2004; 279:9625 - 9633
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311:844 - 847
- Strahl BD, Allis CD. The language of covalent histone modification. Nature 2000; 403:41 - 45
- Sugimoto N, Kitabayashi I, Osano S, Tatsumi Y, Yugawa T, Narisawa-Saito M, et al. Identification of novel human Cdt1-binding proteins by a proteomics approach: proteolytic regulation by APC/CCdh1. Mol Biol Cell 2008; 19:1007 - 1021
- Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, et al. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 2002; 418:994 - 998
- Arentson E, Faloon P, Seo J, Moon E, Studts JM, Fremont DH, et al. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene 2002; 21:1150 - 1158
- Seo J, Chung YS, Sharma GG, Moon E, Burack WR, Pandita TK, et al. Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene 2005; 24:8176 - 8186
- Liontos M, Koutsami M, Sideridou M, Evangelou K, Kletsas D, Levy B, et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res 2007; 67:10899 - 10909
- Mukherjee P, Winter SL, Alexandrow MG. Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol Cell Biol 2010; 30:845 - 856
- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6 and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 2000; 20:8602 - 8612
- Ladenburger EM, Keller C, Knippers R. Identification of a binding region for human origin recognition complex proteins 1 and 2 that coincides with an origin of DNA replication. Mol Cell Biol 2002; 22:1036 - 1048
- Sibani S, Price GB, Zannis-Hadjopoulos M. Decreased origin usage and initiation of DNA replication in haploinsufficient HCT116 Ku80+/− cells. J Cell Sci 2005; 118:3247 - 3261
- Birch JL, Tan BC, Panov KI, Panova TB, Andersen JS, Owen-Hughes TA, et al. FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J 2009; 28:854 - 865