Abstract
DNA damage promotes the activation of a signal transduction cascade referred to as the DNA damage checkpoint. This pathway initiates with the Mec1/ATR kinase, which then phosphorylates the Rad53/Chk2 kinase. Mec1 phosphorylation of Rad53 is then thought to promote Rad53 autophosphorylation, ultimately leading to a fully active Rad53 molecule that can go on to phosphorylate substrates important for DNA damage resistance. In the absence of DNA repair, this checkpoint is eventually down-regulated in a Cdc5-dependent process referred to as checkpoint adaptation. Recently, we showed that overexpression of Cdc5 leads to checkpoint inactivation and loss of the strong electrophoretic shift associated with Rad53 inactivation. Interestingly, this same overexpression did not strongly inhibit Rad53 autophosphorylation activity as measured by the in situ assay (ISA). The ISA involves incubating the re-natured Rad53 protein with γ 32P labeled ATP after electrophoresis and Western blotting. Using a newly identified Rad53 target, we show that despite strong ISA activity, Rad53 does not maintain phosphorylation of this substrate. We hypothesize that, during adaptation, Rad53 may be in a unique state in which it maintains some Mec1 phosphorylation, but does not have the auto-phosphorylations required for full activity towards exogenous substrates.
Introduction
The signaling network that responds to DNA double stranded breaks (DSBs) has been well characterized. Two upstream PI3 kinase-related kinases, called Mec1/ATR and Tel1/ATM, are recruited to the DNA damage site, resulting in their activation. In the case of Mec1/ATR, this activation is mediated, at least in part, by the co-recruitment of a PCNA-related complex called the 9-1-1 complex.Citation1–Citation5 This 9-1-1 complex both directly and indirectly (through the Dpb11 protein) activates Mec1.Citation5–Citation7 Mec1 (and Tel1) then phosphorylate an adaptor molecule, Rad9, which is also recruited to the damage site.Citation8–Citation10 Rad9 phosphorylation promotes its association with the FHA domains of the Rad53 kinase, which is subsequently primed by Mec1/Tel1 phosphorylation. In a poorly defined process, this Rad9-associated and primed Rad53 is then competent to undergo autophosphorylation, resulting in a fully activated kinase.Citation11
Previously, we have shown that, in Saccharomyces cerevisiae, irreparable DSBs do not lead to permanent activation, but instead result in attenuation of the checkpoint signal after approximately six hours.Citation12–Citation14 A loss of function allele of the essential polo kinase CDC5 called cdc5-ad was found to disable this process, leading to persistent checkpoint signaling in response to irreparable breaks.Citation14 Conversely, overexpression of Cdc5, but not Cdc5-ad, leads to premature loss of checkpoint signaling.Citation15 We have suggested that Cdc5-mediated checkpoint inactivation specifically inactivates the step of Rad53 autophosphorylation, since Rad9 phosphorylation and its binding to Rad53 are maintained.Citation15 While it was not possible to uniquely examine Mec1 priming in this scenario, several observations suggested it was maintained. First, Mec1 phosphorylation of Rad9 was largely intact, suggesting that Mec1 kinase activity was not significantly compromised. Second, Rad53 retained a basal electrophoretic shift, consistent with Mec1 priming. Finally, Rad53 activity in vitro, as measured by the in situ assay (ISA), was also largely present despite the fact that the strong electrophoretic shift associated with Rad53 autophosphorylation was lost. Cdc5 overexpression was presumed to inhibit Rad53 activity in vivo based upon both the loss of an electrophoretic shift and the observation that the checkpoint arrest itself was compromised. However, this was difficult to examine directly since we lacked a direct Rad53 substrate showing a robust (and therefore tractable) electrophoretic phospho-shift.
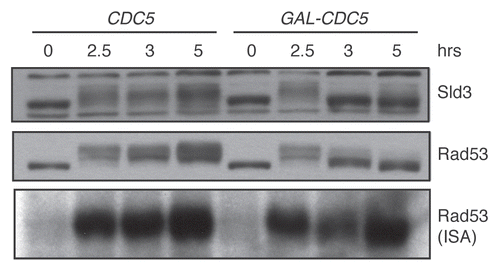
We now examine Rad53 activity in vivo by examining the Rad53-dependent electrophoretic shift of Sld3. Sld3 is an essential protein required for origin firing.Citation16 Our laboratory and the Diffley laboratory recently showed that Sld3 is phosphorylated upon DNA damage in a RAD53-dependent manner.Citation17,Citation18 Moreover, Rad53 directly phosphorylates Sld3 in vitro, suggesting that it is a direct Rad53 target in vivo. The phosphorylation of Sld3 inhibited the ability of Sld3 to promote origin firing. To determine whether Sld3 phosphorylation is lost upon CDC5 overexpression, we examined both Rad53 and Sld3 during a time course of CDC5 overexpression. Cells were damaged for two hours before CDC5 expression was initiated by the addition of galactose. Nocodazole was added with galactose to maintain the G2/M arrest so that our results were not confounded by the cell cycle differences between the CDC5 overexpressing cells (which adapt to the arrest) and control cells lacking the GAL-CDC5 construct. In control samples, both Sld3 and Rad53 phosphorylation were maintained throughout the time course (). In contrast, both Sld3 and Rad53 lost their hyperphosphorylation one hour after galactose addition (the 3 h time point). As seen in our previous experiments, Rad53 ISA activity remained high, despite loss of the hyperphosphorylation normally associated with high Rad53 ISA activity. In fact, Rad53 activity was observed in the faster mobility band, which runs similarly to the mobility of the unphosphorylated, inactive Rad53 protein.Citation15 Thus, loss of Rad53 activity in vivo does not necessarily correlate with loss of ISA activity.
The ISA assay was developed to measure Rad53 autophosphorylation activity.Citation19 Rad53 isolated from DNA-damaged cells has significantly more ISA activity than Rad53 isolated from un-damaged cells.Citation19 Despite the fact that the ISA is widely used, it is unclear what it is actually measuring. Exactly which biological aspects of Rad53 activation are required for this activity is not known, nor is it clear what the phosphorylated substrate is. During this procedure, total protein lysates are renatured on a PVDF membrane. The membrane is subsequently blocked with BSA and incubated with γ32P-labeled ATP for the kinase reaction. It is possible that this BSA serves as a heterologous substrate for the activated kinase. Alternatively, Rad53 could serve as both kinase and substrate. Recombinant Rad53 becomes extensively phosphorylated in bacteria (even more so than in yeast cells after DNA damage), and this material is active against histone H1 in vitro. However, it is only poorly active in an ISA unless it is first partially dephosphorylated in vitro, suggesting that Rad53 is indeed both substrate and enzyme in the ISA and that the hyperphosphorylated Rad53 has no available sites for further autophosphorylation.Citation20 Importantly, bacterially expressed Rad53 is reactive with antibodies against the Mec1/Tel1-phosphorylated residues ([S/T]Q), suggesting that at extremely high concentrations, Rad53 can ectopically hit these sites; therefore, ISA activity of recombinant Rad53 may still require priming phosphorylation of Mec1 sites on Rad53.Citation20 In this sense, the ISA would reflect the second step of checkpoint activation, autophosphorylation. A priori, it must be the case that primed but unautophosphorylated Rad53 is competent, at some level, to phosphorylate another Rad53 molecule, since, after a DSB, the first activated Rad53 molecule has not, by definition, already been Rad53-phosphorylated. Rad53 trans-phosphorylation may simply require basal Rad53 activity coupled to direct juxtaposition of two Rad53 molecules. This could be mediated by multiple Rad53 molecules binding Rad9, followed by direct Rad53-Rad53 interactions. Mec1 phosphorylation of the N terminus of Rad53 promotes Rad53-Rad53 interactions through Rad53's FHA1 domain.Citation21 Thus, Mec1 priming phosphorylation of Rad53 may be the critical modification required for ISA activity. Mec1 (or Tel1) activity is, in fact, required for ISA activity. However, in the absence of these upstream kinases, Rad53 autophosphorylation does not occur, and thus it is impossible to distinguish whether it is the Mec1 phosphorylations, per se, that are required for the ISA. It should be noted that one of the Rad53 autoactivation sites that has been mapped corresponds to the Rad53 activation loop.Citation22 Activation loop phosphorylation increases the activity of many kinases, suggesting that autoactivation may also contribute to ISA activity.Citation23
While we do not know the exact method by which Cdc5 inactivates Rad53, our data are consistent with a model in which Cdc5 phosphorylation of either Rad53 or Rad9 eliminates the ability of Rad9 to promote autophosphorylation of Rad53. This model is consistent with our observation that the ISA activity of Rad53 isolated from Cdc5 overexpressing cells is high, despite the fact that it is neither hyperphosphorylated nor competent to promote Sld3 phosphorylation in vivo. Alternatively, it may be that the hypophosphorylated Rad53 that is observed upon Cdc5 overexpression still retains a subset of the autophosphorylation sites. These sites may be required for in vivo activity, such as localizing Rad53 or promoting substrate interactions, but may not be required for in vitro activity. Interestingly, the human homolog of Cdc5, Plk1, also negatively regulates the mammalian checkpoint pathway.Citation24 This regulation may also target 53BP1 and Chk2, the mammalian equivalents of Rad9 and Rad53 respectively, suggesting that this mode of inhibition is likely conserved.Citation25
Abbreviations
| ISA | = | in situ assay |
| BSA | = | bovine serum albumin |
Figures and Tables
Figure 1 In vivo inhibition of Rad53 activity by CDC5 overexpression. DNA damage was induced at the zero time point in strains yDPT195-1 (cdc13-1 SLD3-3xFLAG::HYG) and yDPT196-1 (cdc13-1 SLD3-3xFLAG::HYG URA3::GalCDC5). After overnight growth in rich media with 2% raffinose, cultures were shifted to 32°C, the non-permissive temperature of the cdc13-1 allele. After two hours, 10 µg/ml nocodazole and 2% galactose, to induce CDC5, were added to the cultures. The samples were collected at the indicated time points and analyzed by western blot and ISA. Sld3 and Rad53 were detected in the western blot with α-FLAG and α-Rad53 (DAB001, gift from the Durocher lab) antibodies, respectively.
Acknowledgements
This work was funded by the National Institutes of Health grant GM59691. J.L.M. was funded by a Ford Foundation Pre-Doctoral Diversity Fellowship. We thank the Durocher lab for kindly sharing their antibody for use in this study.
References
- Bonilla CY, Melo JA, Toczyski DP. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell 2008; 30:267 - 276
- Kondo T, Matsumoto K, Sugimoto K. Role of a complex containing Rad17, Mec3 and Ddc1 in the yeast DNA damage checkpoint pathway. Mol Cell Biol 1999; 19:1136 - 1143
- Majka J, Niedziela-Majka A, Burgers PM. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell 2006; 24:891 - 901
- Melo JA, Cohen J, Toczyski DP. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 2001; 15:2809 - 2821
- Navadgi-Patil VM, Burgers PM. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell 2009; 36:743 - 753
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell 2006; 124:943 - 955
- Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 2008; 22:1478 - 1489
- Emili A. MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell 1998; 2:183 - 189
- Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 1998; 281:272 - 274
- Vialard JE, Gilbert CS, Green CM, Lowndes NF. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J 1998; 17:5679 - 5688
- Gilbert CS, Green CM, Lowndes NF. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell 2001; 8:129 - 136
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998; 94:399 - 409
- Sandell LL, Zakian VA. Loss of a yeast telomere: Arrest, recovery and chromosome loss. Cell 1993; 75:729 - 739
- Toczyski DP, Galgoczy DJ, Hartwell LH. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 1997; 90:1097 - 1106
- Vidanes GM, Sweeney FD, Galicia S, Cheung S, Doyle JP, Durocher D, et al. CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol 2010; 8:e1000286
- Kamimura Y, Tak YS, Sugino A, Araki H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J 2001; 20:2097 - 2107
- Lopez-Mosqueda J, Maas NL, Jonsson ZO, Eli L, Wohlschlegel J, Toczyski DP. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 2010; 467:479 - 483
- Zegerman P, Diffley JFX. Checkpoint dependent inhibition of DNA replication initiation via Sld3 and Dbf4 phosphorylation. Nature 2010; 467:474 - 478
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, et al. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 1999; 18:6561 - 6572
- Ma JL, Lee SJ, Duong JK, Stern DF. Activation of the checkpoint kinase Rad53 by the phosphatidyl inositol kinase-like kinase Mec1. J Biol Chem 2006; 281:3954 - 3963
- Lee SJ, Schwartz MF, Duong JK, Stern DF. Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol Cell Biol 2003; 23:6300 - 6314
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol 2005; 15:1364 - 1375
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 2004; 15:661 - 675
- van Vugt MA, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005; 24:2844 - 2859
- van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1 and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol 2010; 8:e1000287