Abstract
Recent data demonstrated that the aberrant activity of endogenous repetitive elements of the DNA in humans can drive the expression of proto-oncogenes. This article summarizes these results and gives an outlook on the impact of these findings on the pathogenesis and therapy of human cancer.
Repetitive Elementsin Human DNA
A large fraction of the human genome consists of repetitive DNA elements.Citation1 These elements consist of long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), endogenous retroviruses (ERVs) and solitary long terminal repeats (LTRs). Solitary LTRs are in most cases considered to be derived from homologous recombination between both LTRs of a human ERV (HERV)Citation1 integrated in the genome during the course of human evolution. LTRs contain binding sites for transcription factors and for the basal transcription machinery, and it has long been suspected that such ectopically inserted promoters would alter the transcriptional profile of a given cell when activated.Citation1,Citation2 Not much is known about the different scenarios that lead to LTR activation and to what extent such elements have become part of the regulatory machinery of the host genome. Also, the full extent of the functional impact of such repetitive elements on gene regulation and on genome stability in humans is poorly understood.Citation1,Citation3 However, recent data suggest that many such elements are not just nonfunctional “junk” DNA, but have been co-opted by evolution to play important roles in the regulation of gene expression (reviewed in ref. Citation1 and Citation2). For example, endogenous LTRs in normal cells function as alternative promoters regulating tissue-specific gene expression.Citation1,Citation2 This regulation might not only be mediated by LTRs located in the proximity of the usually used original promoter of a gene, but also by long-distance effects on promoters far away from the genomic position of the respective LTR element.Citation4 Currently, it can be assumed that the impact of repetitive elements on gene regulation in human cells has been largely underestimated. Indeed, a recent unbiased analysis of retroelement activity showed that a high number of mRNA transcripts in human cells can be mapped back into repeat regions of the DNA.Citation5
Since the activity of repetitive DNA elements can interfere with normal gene regulation, the activity of such elements has to be tightly controlled. Most repeat elements are epigenetically silenced during embryonic development. One known mechanism to exert such a silencing function is the methylation of repeat element DNA at CG dinucleotides.Citation6 DNA methylation is faithfully maintained during normal development, but is perturbed in cancer cells.Citation7 Human tumors often display epigenetic silencing of tumor suppressor genes,Citation8 but several studies have also reported a global decrease of methylation (reviewed in ref. Citation9), and this includes reports of hypomethylation of repetitive DNA elements.Citation10 However, the impact of altered repeat element methylation patterns on the gene expression pattern of tumor cells and their impact for malignant transformation has not yet been defined.Citation1,Citation11 In particular, it is unclear whether such alterations support the activation of (proto-) oncogenes in human cells.
Lineage-Inappropriate Gene Expression in Classical Hodgkin Lymphoma
In a recent study, we analyzed the mechanisms of classical Hodgkin lymphoma (HL) pathogenesis.Citation12 HL is one of the most frequent lymphomas in western countries. It is characterized by the presence of malignant mono- or multinucleated Hodgkin/Reed-Sternberg (HRS) cells, which usually account for less than 1% of cells in the affected lymph nodes (reviewed in ref. Citation13). The majority of the tumor tissue is composed of normal cells of the immune system, such as B cells, T cells and macrophages, which are suspected to contribute to an immunosuppressive environment around the tumor cells.Citation13 Apart from the unique histopathological presentation of HL-affected lymph nodes, HRS cells show a cellular phenotype that is unusual among human lymphomas.Citation13,Citation14 Although HRS cells in the majority of cases are derived from germinal center B cells, they have nearly completely lost their B-cell phenotype and show an upregulation of non-B genes, i.e., genes of other hematopoietic lineages that are usually not expressed in B cells.Citation14 However, while such expression of non-B lineage genes in the malignant HRS cells is a well known phenomenon, only little is known about their regulation and function. A particular puzzle was the finding that one of the B cell-specific genes that is not expressed is the antigen receptor (BCR),Citation13 which normally provides the major survival signal for mature B cells. It was suggested that EBV infection could play a role in HL pathogenesisCitation13 whereby EBV-encoded genes such as LMP2A would take over such a survival function. This idea was supported by the findings that LMP2A induces a Hodgkin-like phenotype in B cells15 and that EBV can rescue pre-apoptotic B cells from cell death.Citation16
Tumor development is a multiple-step process,Citation17 and the acquisition of a specific malignant phenotype occurs by an extensive selection process which results in the outgrowth of cells that have been reprogrammed to express genes promoting their survival. We therefore explored an alternative concept, i.e., that the acquisition of non-B lineage genes might be caused by such a reprogramming and selection process and that this would also involve the activation of alternative signaling pathways promoting growth and survival of malignant lymphoid cells. In this model, reprogramming and acquisition of non-B genes would be crucial for malignant transformation. Several recent studies support such a concept. In mouse models, PAX5-deficient mature B cells dedifferentiate to early, undifferentiated progenitor cells and are predisposed for the development of aggressive lymphomas.Citation18 Such progenitor cells are characterized by a multilineage gene expression profile, a phenotype also characteristic for HRS cells.Citation13,Citation14 Furthermore, the activation of the PI3 kinase (PI3K) signaling pathway can rescue mature B cells from cell death following loss of BCR signaling.Citation19 Interestingly, PI3K signaling integrates numerous non-B signal transduction pathways and can, for example, be activated by the macrophage-specific receptor for colony-stimulating factor 1 (CSF-1), CSF1R.Citation20 In HL, non-B lineage genes are involved in aberrant cytokine expression and modulation of the microenvironment. For example, the T-cell transcription factor GATA3 regulates IL-13 expression in HRS cells,Citation21 which, in turn, promotes their growth.Citation22 Furthermore, the Th2 cytokine IL-21 modulates the microenvironment surrounding HRS cells via attraction of regulatory T cells.Citation23
The Aberrant Activation of an LTR in HRS Cells Drives Cancer Growth
Two of the genes that are aberrantly expressed in malignant B cell-derived HRS cells are those encoding CSF-1 and its receptor. Our most recent studyCitation12 focused on the regulation of their expression and their function with respect to HL pathogenesis. Both genes are usually not expressed in cells with B-cell origin, but instead their expression is restricted to myeloid cells or, e.g., fibroblasts.Citation24,Citation25 Previously, we showed that the reprogramming process of HRS cells supported CSF1R-upregulation, but the molecular mechanism remained unlcear.Citation26 This was interesting because CSF1R is an oncogenic tyrosine-kinase receptorCitation27 and CSF1R signaling may be involved in lineage switching of hematopoietic cells.Citation28 It was known that CSF-1 was consistently expressed in primary HRS cells,Citation29,Citation30 but data on CSF1R expression in HRS cells were less clear.Citation26,Citation31,Citation32 We detected CSF1R expression as a common feature in HRS cell lines and primary HRS cells.Citation12 Furthermore, aberrantly expressed CSF1R was constitutively activated, most likely by autocrine and paracrine CSF-1 stimulation, and CSF1R signaling was required for HRS cell growth and survival.Citation12 These data strongly supported the idea that non-B genes play a key role in HL biology. However, the expression of these genes, and in particular that of CSF1R, is normally tightly controlled, and the question then arose by which molecular mechanism were they activated in HRS cells?
A key factor for the positive regulation of CSF-1 expression is transcription factor NFκB,Citation33 which is constitutively activated in HRS cells and contributes to their growth and survival.Citation13 Here, we demonstrate that NFκB is a major regulator of CSF-1 also in HRS cells (). Following transfection of HRS cells with the NFκB super-repressor IκBαΔN, an N-terminally truncated, stabilized form of the NFκB inhibitor IκBα, CSF-1 expression was strongly decreased in HRS cell lines (). These data indicate that the aberrant NFκB activity is a major regulator of CSF-1 in HRS cells.
The regulation of CSF1R expression in HRS cells turned out to be more complex.Citation12 CSF1R is normally upregulated during macrophage differentiation, and in B cells it is repressed by the transcription factor PAX5.Citation24,Citation34 Although HRS cells express CSF1R at a level comparable to myeloid cells, we could detect neither open chromatin nor active histone marks at the known CSF1R promoter and FIRE enhancer regions in HRS cells.Citation12 Applying a 5′RACE approach for the detection of unknown 5′-mRNA ends, we instead identified a transcription start site (TSS) in HRS cells located approximately 6.2 kb upstream of the known myeloid promoter. Closer inspection of the sequence around the HRS cell-specific TSS revealed that the CSF1R transcripts in HRS cells originated within an endogenous LTR sequence of the MaLR family called THE1B. Transcriptional activation of the CSF1R LTR promoter was caused by a combination of DNA demethylation of the LTR element together with chronic activation of several transcription factors.Citation12 These included NFκB, AP-1 and GATA factors, all known to be aberrantly activated in HRS cells.Citation13 However, activation of these transcription factors alone was not sufficient to switch on the CSF1R LTR. It turned out that the epigenetic alterations at the CSF1R LTR in HRS cells were linked to the loss of the ETO family member and transcriptional repressor CBFA2T3, which was necessary to maintain LTRs in a repressed state in HRS cells (). Moreover, THE1 LTR activation in HRS cells was not restricted to CSF1R but was widespread in the genome of HRS cells and occurred at many different genomic sites.Citation12 Hence, different genetic and epigenetic alterations had to come together to subvert the tight epigenetic silencing control of THE1 LTR elements. Taken together, it therefore appears as if the HRS cell phenotype is the result of an extensive reprogramming process that leads, on the one hand, to the suppression of lineage-specific gene expression and promotes, on the other hand, to the expression of non-B lineage genes. Moreover, this reprogramming does not only involve an alteration of the chromatin structure of normal promoters but also of endogenous DNA repeat elements.
Pathogenetic Implicationsof Activated Repetitive DNA Elements in Human Malignant Cells
In contrast to normal hematopoietic cells in which the CSF1R LTR region was strongly methylated, its methylation was reduced in various lymphoma and leukemia cell types.Citation12 We detected CSF1R LTR demethylation e.g., in several diffuse large B-cell lymphoma (DLBCL) samples, which, at least in some subtypes, share certain molecular defects with HL,Citation35 but LTR-CSF1R transcripts were undetectable in these lymphomas. Instead, chromatin remodeling appeared to be a key component of LTR activation, as only in HRS cells the CSF1R LTR region was accessible to DNaseI treatment and thus in open chromatin.Citation12 In line with this idea, only HRS cells had lost the transcriptional repressor CBFA2T3, which does not directly alter DNA methylation but recruits factors able to modify chromatin structure.Citation36 These findings indicate that the methylation status of DNA repeat elements does not necessarily reflect their transcriptional activity. Apart from activation of gene expression, aberrant activity of repetitive DNA elements might alter gene expression in other ways. For example, LTR activity might result in the production of non-coding or antisense RNAs or could modify microRNA expression patterns. Even an influence on RNA editing and alterations in nuclear retention have been described.Citation37,Citation38
Our finding naturally raises the question of whether the activation of other families of repetitive elements in humans might have similar consequences. Indeed, in parallel to our work, it has been demonstrated that the aberrant hypomethylation of a long interspersed nuclear element, a class of repetitive elements found in several thousands of copies all over the human genome, is involved in oncogene activation in human bladder cancer.Citation39 These authors studied the regulation of the MET oncogene, which is known to be overexpressed in this cancer type.Citation40 The activation of a LINE-1 promoter within the MET locus resulted in the expression of truncated, but most likely active MET proteins,Citation39 providing a second example of repeat element-driven oncogene activation in human tumors known to date. Interestingly, the analysis of the global LINE-1 methylation status in tissues and cell lines did not predict the methylation status of the LINE-1 MET promoter.Citation39
The studies outlined above raise the possibility that repeat element-driven activation of oncogenes in human malignancies could be wide-spread and has been largely overlooked to date. Although deep-sequencing approaches detect transcripts originating e.g., from alternative promoters, such information is missed in chip-based microarray analyses where repeat DNA is omitted from arrays, and therefore large data sets answering this question will only be available in the near future. Furthermore, it has to be determined whether such transcripts are specific to malignant cells or whether they are found also in benign cell types in the body. This can be expected, since a sigificant percentage of mRNA transcripts in human cells originate in repetitive elements.Citation5 However, if a percentage of such transcripts are specific for malignant cells, this might have major diagnostic, prognostic and therapeutic consequences. For example, we did not detect LTR-CSF1R transcripts in any analyzed normal cell type, whereas they were present in all analyzed HRS cell lines and HL tissue samples.Citation12 Moreover, when examining patterns of LTR-driven transcripts by 3′RACE, it appeared as if each cell line contained a different pattern, hinting that LTR activation across the genome may be stochastic. These transcripts might therefore be useful for the analysis of minimal residual disease in HL patients and might, in general, allow a better “fingerprinting” of a malignant tumor cell clone than it is possible today.
Although several reports show that global alterations of DNA methylation define e.g., patient subgroups with different prognosis,Citation41 we are far away from understanding this finding. Furthermore, it is possible that the aberrant activity of repetitive elements is a driving force behind chromosomal instability and the formation of chromosomal translocations, which are both hallmarks of human malignancies. That this may be the case was highlighted by a recent study demonstrating that the LINE-1 repeat-encoded ORF2 endonuclease is linked to the formation of recurrent chromosomal translocations in human prostate carcinoma cell lines.Citation42 Finally, pharmacological drugs, which alter the epigenetic state of the cells, have now been introduced into the clinic for the treatment of patients suffering from various human malignancies. Although these drugs are effective for the treatment of certain of malignancies, their mechanism of action in patients is only poorly understood.Citation43 Our work shows that treatment of non-Hodgkin cell lines with such pharmacological agents results in the transcriptional activation of CSF1R LTR-driven transcripts.Citation12 Furthermore, such treatment alters the expression of LINE-1-driven transcripts originating in the MET oncogene locusCitation44 and causes LINE-1 demethylation,Citation45,Citation46 and treatment effects are most likely not restricted to malignant cells. The consequences of such effects therefore need to be examined more closely. It also needs to be clarified whether such epigenetic alterations are reversible following cessation of treatment. Moreover, and as already discussed several years ago,Citation47–Citation49 whether the global alteration of epigenotypes predisposes for the activation of oncogenes driven from repetitive elements and for genomic instability and malignant transformation also needs to be studied.
Clinical Implications for the Treatment of Classical Hodgkin Lymphoma
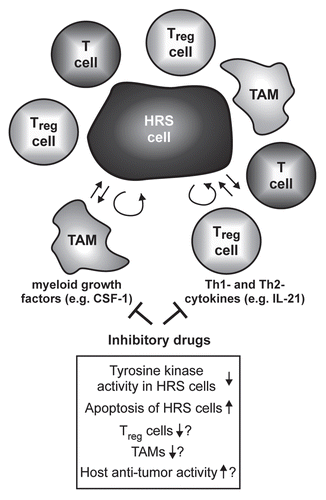
Our workCitation12 might have direct implications for the treatment of HL patients. Although the majority of HL patients can be cured by polychemotherapy with or without radiotherapy, current treatment protocols are accompanied by considerable early and late toxicity, including the induction of treatment-induced secondary malignancies.Citation50 Therefore, it is mandatory to develop new, non-genotoxic targeted treatment strategies for HL patients. In our study, we have provided evidence that inhibition of CSF1R activity blocks growth and induces apoptotic cell death of HRS cell lines.Citation12 Although the pharmacological compounds used in our study are not yet approved for clinical use in patients, other kinase inhibitors have already been succesfully tested for the treatment of a variety of malignancies in patients.Citation51 Some of these substances effectively block CSF1R activity,Citation52 and therefore their effect on patients with HL should be evaluated (). Inhibition of tyrosine-kinase receptor activity including CSF1R might be highly promising for HL treatment for two reasons. First, apart from CSF1R, HRS cells aberrantly express a number of other receptor tyrosine kinases (RTKs) and several of them are activated.Citation53 Multi-tyrosine-kinase inhibitors might therefore not only target CSF1R and associated signaling pathways, but also signaling pathways induced by other RTKs, which could potentiate a possible deleterious effect of such treatment on HRS cells. Secondly, more and more data suggest that the interaction of malignant HRS cells with “innocent” bystander cells surrounding the malignant HRS cells in the affected lymph nodes is central to the growth and survival of these tumor cells.Citation13 Of particular importance for our work is that macrophages might play a central role in this interaction network.Citation54 This is reflected in the fact that a high number of CD68+ tumor-associated macrophages in HL-affected lymph nodes is associated with shortened survival of HL patients.Citation54 Most importantly, although fully differentiated macrophages do not depend on CSF1R signaling, CSF1R activity is indispensable for differentiation and maturation of myeloid precursor cells towards macrophages.Citation20,Citation24 Therefore, inhibition of CSF1R activity as treatment strategy for HL patients might target both the malignant HRS tumor cells and the tumor microenvironment, potentially increasing treatment success.
In summary, our recently published workCitation12 revealed a new mechanism of oncogene activation in human malignant cells. Future work has to define whether similar processes are responsible for oncogene activation in other human malignancies. In addition, altered gene regulation as a result of deregulated repeat activation might be involved in the pathogenesis of other human disorders, e.g., inflammatory and autoimmune diseases.
Figures and Tables
Figure 1 Expression of CSF-1 in HRS cells depends on transcription factor NFκB. Cells of the HRS cell line L428 were transfected with 60 µg of plasmid encoding the NFκB super-repressor IκBαΔN or the respective mock control along with 10 µg pEGFP. Forty-eight hours after transfection, expression of CSF-1 was analyzed in GFP-positive cells by intracellular flow cytometry (upper part, antibody to CSF-1, MAB216; R&D Systems). IC, isotype control (MAB003; R&D Systems). Expression of IκBαΔN in L428 cells was verified by western blotting (lower part, antibody to IκBα, sc-371; Santa Cruz). Note that L428 cells lack endogenous IκBα expression due to a deleterious mutation of the NFKBIA gene.Citation55 Reh and Namalwa non-Hodgkin cell lines and expression of β-actin (antibody to β-actin, A5316, Sigma-Aldrich) were analyzed as controls.
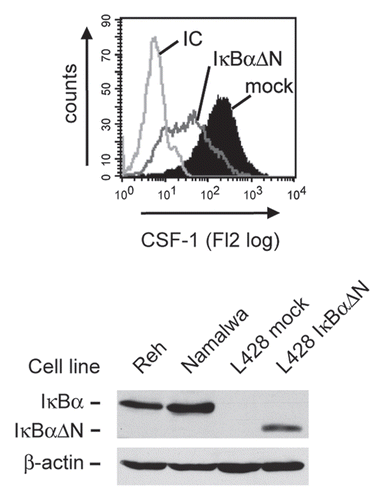
Figure 2 Model of aberrant endogenous LTR activation in Hodgkin lymphoma cells. Upper panel, In normal mature B cells, DNA upstream of the CSF1R locus including the CSF1R LTR is organized in compact heterochromatin that carries inactive histone marks such as histone H3 K9 trimethylation and methylated DNA. This recruits methyl binding proteins (MeCP), polycomb complexes (PcG) and heterochromatin protein (HP1).Citation56 The heterochromatic state of the LTR is maintained by the CBFA2T3 silencing complex. The normal CSF1R promoter is repressed by the transcription factor PAX5 which blocks the PU.1-mediated recruitment of RNA polymerase II.Citation34 Lower panel, in HRS cells, PU.1 and CBFA2T3 expression is lost, silent chromatin is not maintained and DNA is demethylated. In addition, inducible transcription factors such as NFκB and AP-1 are chronically activated and bind to the LTR. Chromatin is remodeled, leading to the recruitment of RNA polymerase II to the LTR and active transcription bypassing the normal promoter.
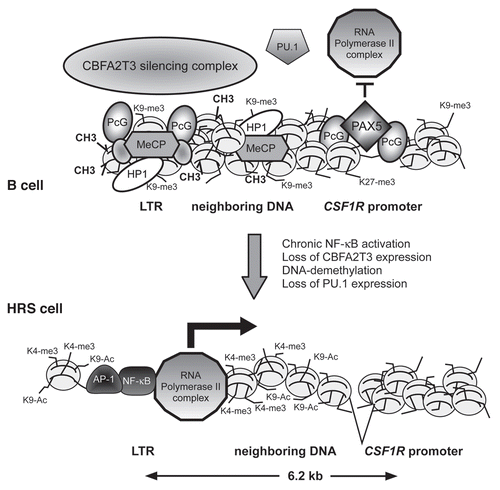
Figure 3 Simplified model of the influence of non-B cytokines on the microenvironment of HRS cells and possible consequences of their inhibition. The malignant HRS cells are surrounded by immune bystander cells like CD4+ T cells, regulatory T cells (Treg) or macrophages (among these tumor-associated macrophages, TAM). The interaction of these cells is supposed to support growth and survival of the tumor cells. Cytokines like the Th2 cytokine IL-21 or the myeloid growth factor CSF-1 are produced by the HRS cellsCitation12,Citation23 and act not only in an autocrine fashion but also paracrine on T cells and TAM. Inhibitory drugs targeting these cytokines and their associated signaling pathways might therefore not only induce growth arrest and apoptosis of the HRS cells, but also disrupt the tumor microenvironment, thereby targeting HRS cells indirectly.
Acknowledgements
We apologize to colleagues whose articles could not be cited because of space limitations. We thank Martin Janz for critical reading of the manuscript. Work of the authors described in this article was supported in part by grants from the Deutsche Forschungsgemeinschaft, the Berliner Krebsgesellschaft, the Wilhelm Sander-Stiftung, Leukaemia and Lymphoma Research, Cancer Research UK and Yorkshire Cancer Research.
References
- Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu Rev Genet 2008; 42:709 - 732
- Cohen CJ, Lock WM, Mager DL. Endogenous retroviral LTRs as promoters for human genes: A critical assessment. Gene 2009; 448:105 - 114
- Romanish MT, Cohen CJ, Mager DL. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin Cancer Biol 2010; 20:246 - 253
- Pi W, Zhu X, Wu M, Wang Y, Fulzele S, Eroglu A, et al. Long-range function of an intergenic retrotransposon. Proc Natl Acad Sci USA 2010; 107:12992 - 12997
- Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 2009; 41:563 - 571
- Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci 2008; 65:3329 - 3347
- Ehrlich M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002; 21:5400 - 5413
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 428
- Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics 2009; 1:239 - 259
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta 2007; 1775:138 - 162
- Druker R, Whitelaw E. Retrotransposon-derived elements in the mammalian genome: a potential source of disease. J Inherit Metab Dis 2004; 27:319 - 330
- Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med 2010; 16:571 - 579
- Küppers R. The biology of Hodgkin's lymphoma. Nat Rev Cancer 2009; 9:15 - 27
- Janz M, Dörken B, Mathas S. Reprogramming of B lymphoid cells in human lymphoma pathogenesis. Cell Cycle 2006; 5:1057 - 1061
- Portis T, Dyck P, Longnecker R. Epstein-Barr Virus (EBV) LMP2A induces alterations in gene transcription similar to those observed in Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003; 102:4166 - 4178
- Mancao C, Altmann M, Jungnickel B, Hammerschmidt W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005; 106:4339 - 4344
- Greaves M. Cancer stem cells: Back to Darwin?. Semin Cancer Biol 2010; 20:65 - 70
- Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 2007; 449:473 - 477
- Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009; 139:573 - 576
- Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol 2004; 14:628 - 638
- Stanelle J, Döring C, Hansmann ML, Küppers R. Mechanisms of aberrant GATA-3 expression in classical Hodgkin lymphoma and its consequences for the cytokine profile of Hodgkin and Reed/Sternberg cells. Blood 2010; 116:4202 - 4211; http://dx.doi.org/10.1182/blood-2011-01-265827
- Kapp U, Yeh WC, Patterson B, Elia AJ, Kagi D, Ho A, et al. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J Exp Med 1999; 189:1939 - 1946
- Lamprecht B, Kreher S, Anagnostopoulos I, Jöhrens K, Monteleone G, Jundt F, et al. Aberrant expression of the Th2 cytokine IL-21 in Hodgkin lymphoma cells regulates STAT3 signaling and attracts Treg cells via regulation of MIP-3alpha. Blood 2008; 112:3339 - 3347
- Bonifer C, Hume DA. The transcriptional regulation of the colony-stimulating factor 1 receptor (csf1r) gene during hematopoiesis. Front Biosci 2008; 13:549 - 560
- Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008; 8:533 - 544
- Mathas S, Janz M, Hummel F, Hummel M, Wollert-Wulf B, Lusatis S, et al. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol 2006; 7:207 - 215
- Roussel MF, Sherr CJ. Oncogenic potential of the c-FMS proto-oncogene (CSF-1 receptor). Cell Cycle 2003; 2:5 - 6
- Borzillo GV, Ashmun RA, Sherr CJ. Macrophage lineage switching of murine early pre-B lymphoid cells expressing transduced fms genes. Mol Cell Biol 1990; 10:2703 - 2714
- Janowska-Wieczorek A, Belch AR, Jacobs A, Bowen D, Padua RA, Paietta E, et al. Increased circulating colony-stimulating factor-1 in patients with preleukemia, leukemia and lymphoid malignancies. Blood 1991; 77:1796 - 1803
- Moreau A, Praloran V, Berrada L, Coupey L, Gaillard F. Immunohistochemical detection of cells positive for colony-stimulating factor 1 in lymph nodes from reactive lymphadenitis, and Hodgkin's disease. Leukemia 1992; 6:126 - 130
- Paietta E, Racevskis J, Stanley ER, Andreeff M, Papenhausen P, Wiernik PH. Expression of the macrophage growth factor, CSF-1 and its receptor c-fms by a Hodgkin's disease-derived cell line and its variants. Cancer Res 1990; 50:2049 - 2055
- Hsu PL, Lin YC, Hsu SM. Expression of macrophage colony-stimulating factor (M-CSF) in two Hodgkin's Reed-Sternberg (H-RS) cell lines, HDLM-1 and KM-H2 and in H-RS cells in tissues. Int J Hematol 1991; 54:315 - 326
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, et al. IKKalpha, IKKbeta and NEMO/IKKgamma are each required for the NFkappaB-mediated inflammatory response program. J Biol Chem 2002; 277:45129 - 45140
- Tagoh H, Ingram R, Wilson N, Salvagiotto G, Warren AJ, Clarke D, et al. The mechanism of repression of the myeloid-specific c-fms gene by Pax5 during B lineage restriction. EMBO J 2006; 25:1070 - 1080
- Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med 2010; 362:1417 - 1429
- Hug BA, Lazar MA. ETO interacting proteins. Oncogene 2004; 23:4270 - 4274
- Chen LL, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. EMBO J 2008; 27:1694 - 1705
- Chen LL, Carmichael GG. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell 2009; 35:467 - 478
- Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, et al. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 2010; 6:e1000917
- Cheng HL, Trink B, Tzai TS, Liu HS, Chan SH, Ho CL, et al. Overexpression of c-met as a prognostic indicator for transitional cell carcinoma of the urinary bladder: a comparison with p53 nuclear accumulation. J Clin Oncol 2002; 20:1544 - 1550
- Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010; 17:13 - 27
- Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009; 139:1069 - 1083
- Bonifer C, Bowen DT. Epigenetic mechanisms regulating normal and malignant haematopoiesis: new therapeutic targets for clinical medicine. Expert Rev Mol Med 2010; 12:6
- Weber B, Kimhi S, Howard G, Eden A, Lyko F. Demethylation of a LINE-1 antisense promoter in the cMet locus impairs Met signalling through induction of illegitimate transcription. Oncogene 2010; 29:5775 - 5784; http://dx.doi.org/10.1038/onc.2010.227
- Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32:38
- Yang AS, Doshi KD, Choi SW, Mason JB, Mannari RK, Gharybian V, et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res 2006; 66:5495 - 5503
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300:455
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300:489 - 492
- Yang AS, Estecio MR, Garcia-Manero G, Kantarjian HM, Issa JP. Comment on “Chromosomal instability and tumors promoted by DNA hypomethylation” and “Induction of tumors in nice by genomic hypomethylation”. Science 2003; 302:1153
- Diehl V, Engert A, Re D. New strategies for the treatment of advanced-stage Hodgkin's lymphoma. Hematol Oncol Clin North Am 2007; 21:897 - 914
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9:28 - 39
- Kumar R, Crouthamel MC, Rominger DH, Gontarek RR, Tummino PJ, Levin RA, et al. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer 2009; 101:1717 - 1723
- Renné C, Willenbrock K, Küppers R, Hansmann ML, Bräuninger A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood 2005; 105:4051 - 4059
- Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med 2010; 362:875 - 885
- Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, et al. Overexpression of IkappaB alpha without inhibition of NFkappaB activity and mutations in the IkappaBalpha gene in Reed-Sternberg cells. Blood 1999; 94:3129 - 3134
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat Rev Genet 2009; 10:295 - 304