Abstract
Comment on: Thomas Y, et al. Cell Cycle 2010; 9:4338–50.
Considered the “engines” of cell cycle progression, cyclin-dependent kinases (CDKs) phosphorylate myriad downstream substrates to promote cell growth, replication and division. Tight control of CDK activity results from, among other mechanisms, the opposing actions of inhibitory kinases (Wee1 and Mik1) and activating phosphatases. Cdc25 phosphatases are essential components of this process, reversing inhibitory phosphorylation of CDKs during key cell cycle transitions and peaking in their activity as cell division nears its mitotic end.
Although the three mammalian Cdc25 paralogs—Cdc25A, Cdc25B and Cdc25C—are regulated by multiple mechanisms, degradation of Cdc25A and Cdc25B by the ubiquitin-proteasome system remains a major method of regulation during the cell cycle. interestingly, Cdc25 genes are considered proto-oncogenes whose excessive activity accelerates proliferation, but the increased levels of Cdc25A and Cdc25B observed in tumors do not generally occur as a consequence of amplification or mutations that lead to overt overexpression. instead, elevated Cdc25 levels result from their stabilization and persistence during cell cycle periods when they should be absent or degraded.Citation1
The functional characteristics that distinguish the Cdc25 paralogs have been increasingly investigated. While the mouse knockout model of Cdc25A displays embryonic lethality, deletion of either Cdc25B or Cdc25C imparts little phenotypic outcome at both the organismal and cellular levels.Citation2 In fact, cells derived from mice lacking both Cdc25B and Cdc25C do not show significant defects in their cell cycles or DNA damage responses, with these normal phenotypes perhaps indicating redundancy in their functions and substrates. In contrast to these loss-of-function studies, examining the aberrant persistence of these proteins may reveal more about their particular roles and elucidate more subtle mechanisms of their regulation. Studies investigating the specific conditions for Cdc25A degradation mediated by the Anaphase Promoting Complex/Cyclosome (APC/CCdh1) or the SCFβTrCP ubiquitin ligases have led to a greater understanding of the delicate balance of Cdc25A levels across the cell cycleCitation3 (). Now, by confirming and extending the long-postulated notion that Cdc25B, like Cdc25A, is degraded by the ubiquitin-proteasome system via the F-box protein βTrCP,Citation4 the work of Thomas et al. In a previous issue of Cell Cycle furthered this discussion of distinct roles within the Cdc25 family.Citation5
In their report, the authors investigate the unique features of Cdc25B activity during mitosis, showing βTrCP-dependent degradation at the metphase-anaphase transition—in surprising contrast to the nearly simultaneous removal of Cdc25A through APC/CCdh1—and raise intriguing questions about novel mechanisms regulating βTrCP-mediated ubiquitylation. In S phase, when the activity of CDKs must be maintained at levels low enough to be permissive for DNA replication, Cdc25A levels are moderated by βTrCP. Following S-phase, CDK activity rises, inhibiting replication (thereby preventing reduplication) and promoting mitotic events.Citation3 In response to DNA damage in S and G2, βTrCP-dependent degradation of Cdc25A is enhanced. These interphase and DNA damage-responsive interactions between Cdc25A and βTrCP require phosphorylation by a number of kinases—one priming (Chk1),Citation3 and others processive (CKIα, GSK3β and Nek11),Citation6,Citation7 with binding dependent upon the final phosphorylation within the protein's βTrCP degron, an inverted variant of the classical DSGxxS motif (STDSG).
The many characterized substrates of βTrCP share some version of this DSGxxS phosphodegron, sometimes with phosphomimetic substitution, but Thomas et al. instead show that Cdc25B uniquely lacks any phosphorylable residue in its degron sequence (DDGFvD). Since regulation of the timely recognition of substrates by βTrCP famously follows a phosphorylation within this motif, how might Cdc25B ubiquitylation be controlled?
The authors speculate that regulation of Cdc25B degradation in the absence of a phosphorylable degron may be due to modification of βTrCP itself, or it may represent a kind of competition for βTrCP, in which occupation of its substrate recognition site by phosphorylated substrates (which are likely to bind βTrCP with high affinity) allows Cdc25B to accumulate, but permits its degradation at the end of mitosis after other substrates have been eliminated. The abundance of βTrCP in the cell and its diverse substrate targets argue that perhaps another mechanism might exist. One way to counteract a ‘constitutively active’ degron would be to oppose ubiquitylation in a regulated manner, namely using a deubiquitylating enzyme (DUB). Thus, it is possible that a yet undiscovered DUB stabilizes Cdc25B at the G2/M transition. For example, recent evidence indicates that Dub3 (also called USP17) positively regulates Cdc25A levels and has been found to be elevated in human breast cancer cell lines in which Cdc25A is stabilized.Citation8 Dub3 may therefore contribute to tumorigenesis by inappropriately stabilizing Cdc25A and, conceivably, other substrates.
High expression of exogenous Cdc25B is known to induce mitotic catastrophe, and overexpression of Cdc25B in S phase results in centrosome overduplication.Citation9 in their report, Thomas et al. show that a stabilized Cdc25B mutant (unable to bind βTrCP) yields discrete cellular consequences, including a delay in mitotic exit, diverse chromosomal and spindle defects and the appearance of micronuclei that accrue in subsequent mitoses. Therefore, the regulation of Cdc25B turnover in the absence of a phosphorylable degron is a subject of interest for future studies of Cdc25 phosphatase activity during the cell cycle, and exploration of alternative regulatory mechanisms may provide a greater overall understanding of regulated proteolysis.
Figures and Tables
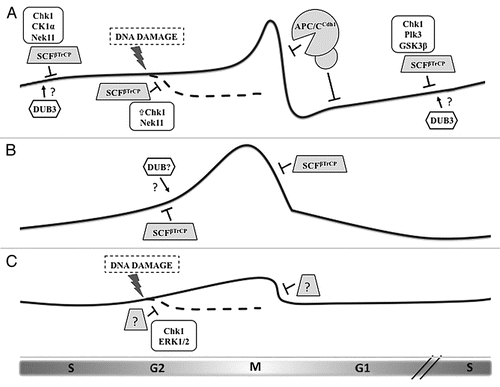
Figure 1 Regulation of the three CDC25 paralogs during the cell cycle by ubiquitin-mediated degradation. (A) Cdc25A protein levels begin to rise in late G1 and remain modest during S phase and early G2 by the action of the ubiquitin ligase SCFβTrCP, whose recognition of Cdc25A depends upon phosphorylation by a number of kinases. DNA damage induction of Chk1 activity increases the priming phosphorylation of Cdc25A, leading to greater βTrCP-induced degradation. At the end of mitosis and through G1, Cdc25A is eliminated via the APC/CCdh1 ubiquitin ligase. Dub3 has been shown to stabilize Cdc25A by deubiquitylation, although the precise timing and conditions for its activity remain unknown. (B) Cdc25B levels are also moderated by βTrCP, but in a phosphorylation-independent manner. Rapid destruction of Cdc25B at the metaphase-anaphase transition is mediated by βTrCP. Since the degron of Cdc25B mimics constitutive phosphorylation, we hypothesize that another mechanism counteracts βTrCP-mediated ubiquitylation during its rise in G2, possibly the activity of a deubiquitylating enzyme (DUB). (C) Cdc25C is the least-studied protein of the family, and its regulation is determined mostly by phosphorylation events that regulate its phosphatase activity and localization. Evidence exists that Cdc25C is ubiquitylated and degraded following certain forms of G2 arrest, but, although this event requires Chk1 and ERK1/2, the contributing ubiquitin ligase is unknown.
References
- Löffler H, et al. Oncogene 2003; 22:8063 - 8071
- Guardavaccaro, et al. Mol Cell 2006; 22:1 - 4
- Frescas D, et al. Nat Rev Cancer 2008; 8:438 - 449
- Kanemori Y, et al. Proc Natl Acad Sci USA 2005; 102:6279 - 6284
- Thomas Y, et al. Cell Cycle 2010; 9:4338 - 4350
- Melixetian M, et al. Nat Cell Biol 2009; 11:1247 - 1253
- Honaker Y, et al. Oncogene 2010; 29:3324 - 3334
- Pereg Y, et al. Nat Cell Biol 2010; 12:400 - 406
- Boutros R, et al. Cancer Res 2007; 67:11557 - 11564