Abstract
Studies in budding yeast suggest the protein kinase Rad53 plays novel roles in controlling initiation of DNA replication and in maintaining cellular histone levels, and these roles are independent of Rad53-mediated regulation of the checkpoint and of nucleotide levels. In order to elucidate the role of Rad53 in replication initiation, we isolated a novel allele of RAD53, rad53-rep,that separates the checkpoint function of RAD53 from the DNA replication function. rad53-rep mutants display a chromosome loss phenotype that is suppressed by increased origin dosage, providing further evidence that Rad53 plays a role in the initiation of DNA replication. Deletion of the major histone H3-H4 pair suppresses rad53-rep-cdc7-1 synthetic lethality, suggesting Rad53's functions in degradation of excess cellular histone and in replication initiation are related. Rad53-rep is active as a protein kinase yet fails to interact with origins of replication and like the rad53D mutant, the rad53-rep mutant accumulates excess soluble histones, and it is sensitive to histone dosage. In contrast, a checkpoint defective allele of RAD53 with mutations in both FHA domains, binds origins, and growth of a rad53-FHA mutant is unaffected by histone dosage. Based on these observations, we hypothesize that the origin binding and the histone degradation activities of Rad53 are central to its function in DNA replication and are independent of its checkpoint functions. We propose a model in which Rad53 acts as a "nucleosome buffer," interacting with origins of replication to prevent the binding of excess histones to origin DNA and to maintain proper chromatin configuration.
Introduction
Eukaryotic cells have evolved elaborate mechanisms to ensure that the entire genome is replicated once and only once per cell cycle and to ensure that replication is complete before cellular division in M phase. Components of DNA replication machinery assemble on chromatin in a highly regulated manner that is driven by cell cycle progression.Citation1,Citation2 In budding yeast Saccharomyces cerevisiae the origin recognition complex (ORC) is bound to origin DNA, a site of DNA replication initiation, throughout the cell cycle. Early in G1 phase, Cdc6 and Cdt1 recruit the MCM complex to the ORC-associated origin to form the pre-RC.Citation3,Citation4 The MCM complex, a heterohexameric ring consisting of subunits Mcm2-7, is required for the initiation and elongation steps of DNA replication,Citation5 and it is the putative replicative helicase.Citation6
Pre-RCs are assembled at origins of replication throughout the yeast genome, marking potential sites for the initiation of DNA replication. A subset of these complexes is activated in a temporal manner throughout S phase.Citation7 Activation of pre-RCs depends on two protein kinases, cyclin dependent protein kinase (CDK; Cdk1-Clb) and Dbf4-dependent kinase (DDK; Cdc7-Dbf4).Citation2 CDK phosphorylates DNA replication proteins Sld2 and Sld3,Citation8,Citation9 and the MCM complex is the target of DDK kinase activity in promoting pre-RC activation.Citation10,Citation11 Specific mutations in the MCM complex bypass the requirement for DDK in replication initiation.Citation12,Citation13 The P83L mutation of the mcm5-bob1 bypass allele induces a conformational change in the MCM complex which mimics DDK phosphorylation and subsequent activation of the MCM complex in wild type cells.Citation14,Citation15 Deletion of N-terminal residues of MCM4 (mcm4Δ74-174), which are thought to play an inhibitory role in DNA replication initiation, also promotes bypass of DDK.Citation13
Eukaryotic cellular checkpoint responses monitor the completion and predetermined order of cell cycle events such as DNA replication and mitosis. When these events are disrupted, checkpoints halt cell cycle progression until the earlier process is complete.Citation16–Citation18 In S. cerevisiae the Rad53 protein kinase plays important roles in the G1, S and G2 phases of the cell cycle. Signals from DNA damage or stalled replication forks activate the Mec1 kinase which activates the Rad53 kinase, and activated Rad53 phosphorylates effectors of the checkpoint signal.Citation19,Citation20 This cascade of signaling events ultimately causes a cellular checkpoint response including cell cycle delay, expression of DNA repair proteins, translesion synthesis, and replication fork stabilization.Citation17,Citation21,Citation22
In addition to its function in transducing and amplifying signal in the DNA damage and DNA replication checkpoints, an important function of Rad53 is to monitor replication fork stability at sites of replication. rad53 mutants form aberrant DNA structures at replication forks after treatment with the nucleotide limiting factor hydroxyurea (HU)Citation23,Citation24 or if a replication fork encounters DNA damage.Citation25,Citation26 Formation of these aberrant structures is lethal to the cell, thus a major function of Rad53 and other checkpoint proteins is to stabilize paused replication forks after the active replication fork encounters damaged DNA or other replication perturbations.
The Rad53 protein consists of a kinase domain flanked by two forkhead homology-associated (FHA) domains, FHA1 and FHA2.Citation27 These well-conserved protein domains facilitate interaction of Rad53 with phosphorylated proteins.Citation28 In particular, the Rad53 FHA domains facilitate interaction of Rad53 with upstream sensor and mediator proteins and with downstream Rad53-regulated proteins during the checkpoint response.Citation29–Citation34 Rad53 FHA domains are also necessary for Rad53 oligomer formation and subsequent Rad53 activation after DNA damage.Citation35 The FHA domains of Rad53 are essential for Rad53's role in the DNA damage and replication checkpoints; mutation of both FHA1 and FHA2 domains of Rad53 eliminates almost all checkpoint responses.Citation32
Rad53 kinase also has an essential function in upregulating levels of nucleotides during DNA replication.Citation36,Citation37 The essential function of Rad53 is dependent on its kinase activity,Citation38,Citation39 but independent of the Rad53 FHA domains.Citation40 Overexpression of RNR1, which encodes large subunit of ribonucleotide reductase (RNR) or deletion of the RNR inhibitor SML1 (sml1Δ) suppresses the lethality of a mec1Δ or a rad53Δ single mutant.Citation41,Citation42
Rad53 has other roles in the cell cycle independent of its essential and checkpoint functions. Rad53 maintains cellular histone levels by phosphorylating excess non-nucleosomal histones, which are then ubiquitylated and degraded by the proteasome.Citation43,Citation44 rad53Δ but not mec1Δ mutants are sensitive to overexpressed histone H3, and deleting the major H3–H4 gene pair partially suppresses the DNA damage sensitivity, chromosome loss and slow growth phenotypes of rad53Δ cells,Citation43 suggesting that this housekeeping function of Rad53 is independent of its functions in the checkpoints.
Certain alleles of RAD53 and deletion of RAD53 are synthetically lethal with the temperature sensitive mutation cdc7-1, suggesting that Rad53 functions during DNA replication.Citation45 This replication function is independent of Rad53's essential function and the known checkpoint functions of RAD53, because mutations of other genes in checkpoint response pathways are not synthetically lethal with cdc7-1, and because mutation of the RAD53 FHA domains, which are required for Rad53 checkpoint function has no effect on viability in a cdc7-1 mutant background.Citation45,Citation46 mcm5-bob1 bypass of CDC7 is dependent on RAD53, and mcm5-bob1 has no effect on cdc7-rad53 synthetic lethality. rad53Δ mutants display a minichromosome loss phenotype which is rescued by addition of multiple origins of replication to the minichromosome,Citation46 suggesting Rad53 is involved in the initiation of replication. Deletion of the major histone H3–H4 gene pair suppresses synthetic lethality of rad53Δ with cdc7-1,Citation46 therefore Rad53 function in regulating DNA replication initiation may be related to its function in regulating cellular histone levels.Citation43,Citation44
We report isolation of a novel separation of function allele of RAD53, rad53-rep. The rad53-rep mutant is proficient in the DNA checkpoint but is synthetically lethal with cdc7-1. Characterization of the rad53-rep mutant suggests that the role of Rad53 in the initiation of DNA replication is dependent on Rad53 kinase activity and association of Rad53p with origins of replication. Reducing histone H3–H4 gene dosage suppresses the replication defects of a rad53-rep mutant. More importantly, we show that like the rad53Δ mutant,Citation43 the rad53-rep mutant is sensitive to elevated histone levels, while a checkpoint-defective rad53 mutant with mutations in both FHA domains is not. Finally, like a rad53Δ mutant,Citation43,Citation44 the rad53-rep mutant also accumulate excess histones that are not incorporated into chromatin. Thus, the roles of Rad53 in replication initiation and in histone homeostasis are genetically separable from Rad53's role in the checkpoint response. Our work supports a model in which Rad53 positively regulates DNA replication initiation by indirectly maintaining proper chromatin structure at origins of replication.
Results
rad53-rep is a checkpoint proficient, replication defective allele of RAD53.
In order to more clearly define the role of Rad53 in DNA replication, we performed a genetic screen to isolate alleles of RAD53 that had retained DNA damage checkpoint activity but had lost DNA replication activity. Because Rad53 kinase activity is necessary for both viability in a cdc7-1 mutant and for suppression by mcm5-bob1,Citation46 we specifically generated mutations in the RAD53 kinase domain by amplifying nucleotides 476–1,488 of RAD53 by PCR under mutagenic conditions.
A cdc7-1 rad53Δ sml1Δ strain with wild type CDC7 expressed from a URA3 plasmid was co-transformed with a pool of randomly mutated RAD53 kinase domain fragments generated by mutagenic PCR and with a linearized LEU2 RAD53 plasmid. Homologous recombination occurring within the cell resulted in a LEU2 RAD53 plasmid in which the RAD53 kinase domain was randomly mutated. We then screened approximately 1,500 Leu+ transformants for checkpoint activity by selecting HU resistant (HUR) colonies. To isolate alleles of RAD53 that were checkpoint proficient but defective in replication function, HUR colonies were tested for viability in a cdc7-1 mutant background by screening for resistance to 5-FOA, which counterselects for the URA3 CDC7 plasmid. Cells containing a LEU2 RAD53 plasmid on which the RAD53 allele was proficient in replication would not be dependent on the URA3 CDC7 plasmid for growth and would grow on media containing 5-FOA. Transformants containing a LEU2 plasmid on which the RAD53 allele was defective in replication would be dependent on the URA3 CDC7 plasmid for viability, and these transformants would not be able to grow in the presence of 5-FOA. Of the 112 HUR transformants screened, one was also sensitive to 5-FOA, and the mutant allele of RAD53 on the plasmid was designated rad53-rep (). rad53-rep contains seven point mutations: S175Y; N189D; S198L; K213R; H426R; K459E and D526N. Mutation of histidine 426 to arginine (rad53 H426R) is sufficient for the separation of function phenotype ().
To determine whether rad53-rep was dominant or recessive, a copy of wild type RAD53 or rad53-rep was integrated at the URA3 locus of different cdc7-1 sml1Δ strains (). Wild type RAD53 integrated at the URA3 locus complements the replication and checkpoint defects in a rad53Δ strain (Holzen TM and Sclafani RA, unpublished data). Because RAD53 expressed from the URA3 locus is fully functional, a URA3-RAD53 cdc7-1 rad53Δ sml1Δ strain is not dependent on a TRP1 RAD53 plasmid for growth, as illustrated by its ability to grow in the presence of 5-fluoroanthranillic acid (5-FAA), which counterselects for the TRP1 plasmid expressing wild type RAD53. Because rad53-rep is synthetically lethal with cdc7-1, cdc7-1 rad53Δ sml1Δ strains with rad53-rep integrated at the URA3 locus are dependent on a plasmid-borne copy of wild type RAD53 for growth. cdc7-1 strains that express rad53-rep from the URA3 locus and an endogenous copy of wild type RAD53 are not dependent on the TRP1 RAD53 plasmid for growth, thus the DNA replication defect of rad53-rep is recessive.
Rad53 kinase activity is necessary for mcm5-bob1 to bypass the requirement for DDK for replication initiation.Citation46 We next determined whether rad53-rep promoted mcm5-bob1 bypass of the cdc7-1 mutations. Wild type RAD53, rad53-rep or an empty URA3 vector was integrated at the URA3 locus of a cdc7-1 mcm5-bob1 rad53Δ sml1Δ strain expressing wild type RAD53 on a TRP1 plasmid (). These Ura+ transformants are viable on YEPD media at 30°C, the restrictive temperature for the temperature sensitive cdc7-1 mutation, because the mcm5-bob1 mutation bypasses the requirement for CDC7 function for replication initiation. The RAD53 cdc7-1 mcm5-bob1 strain is also viable on 5-FAA at 30°C because it is not dependent on the TRP1 RAD53 plasmid for mcm5-bob1 suppression of cdc7-1. mcm5-bob1 bypass of the cdc7-1 mutation is dependent on RAD53,Citation46 thus the rad53Δ cdc7-1 mcm5-bob1 mutant cannot grow in the absence of the TRP1 RAD53 plasmid. The rad53-rep cdc7-1 mcm5-bob1 strain resembles the rad53Δ strain; it is not viable on media containing 5-FAA, thus rad53-rep does not support mcm5-bob1 bypass of DDK. We used a similar plasmid shuffle assay to test whether the rad53 H426R mutation promotes mcm4Δ74-174 bypass of DDK.Citation13 A mcm4Δ74-174 construct was integrated at the MCM4 gene in a URA3-rad53 H426R cdc7-1 rad53Δ sml1Δ strain. The rad53 H426R cdc7-1 mcm4Δ74-174 mutant, like the untransformed rad53 H426R cdc7-1 MCM4+ strain, is unable to lose the TRP1 RAD53 plasmid at 30°C (Holzen TM and Sclafani RA, unpublished data). We conclude that rad53-rep and rad53 H426R suppress mutations in the MCM complex that bypass DDK and that bypass mutations in the MCM complex do not suppress the synthetic lethality of rad53-rep or rad53 H426R with cdc7-1.
Unlike the rad53Δ mutant, the rad53-rep mutant is not dependent on mutation of SML1 or on a high-copy plasmid overexpressing RNR1, thus rad53-rep is proficient in Rad53's essential function of regulating nucleotide levels (Holzen TM and Sclafani RA, unpublished data). Therefore, although rad53-rep performs the essential function of Rad53 in regulating cellular nucleotide levels, it is defective in Rad53's function in DNA replication.
The DNA replication and the DNA damage checkpoints are intact in rad53-rep mutants.
Rad53 promotes cell survival after replication stress by stabilizing stalled replication forks,Citation23,Citation24 therefore we determined whether rad53-rep mutants can recover from HU-induced cell cycle arrest (). Cultures of URA3-RAD53 sml1Δ, rad53Δ sml1Δ, URA3-rad53-rep sml1Δ and URA3-rad53 H426R sml1Δ strains were incubated in YEPD containing HU for increasing time periods and then transferred to YEPD without HU to release cells from HU-induced arrest. rad53-rep sml1Δ and rad53 H426R sml1Δ mutants retain wild type levels of viability after exposure to HU.
Because Rad53 also plays a central role in the checkpoint responses throughout the cell cycle,Citation17,Citation41,Citation47 we also investigated whether rad53-rep mutants activated checkpoints after exposure to ultraviolet light (), the DNA alkylating agent MMS, and the DNA intercalating agent zeocin (). In all cases, the rad53-rep mutant resembled wild type regarding resistance to these DNA damaging agents. rad53-rep showed wild type levels of viability after exposure to HU, UV and genotoxic agents, thus we conclude that the DNA damage and the S phase checkpoints of a rad53-rep mutant are intact.
Activation of the S phase checkpoint induces Rad53 kinase activity in a rad53-rep mutant.
During the DNA replication checkpoint response, the Mec1/ATR sensor phosphorylates the adaptor protein Mrc1, which in turn activates Rad53.Citation29,Citation48 Although cell viability assays have shown that the rad53-rep mutant appears to be proficient in the G1, G2 and S-phase checkpoints, we used western blotting ( and C) and the in situ autophosphorylation assay (ISA; ; reviewed in ref. Citation49) to examine autophosphorylation of Rad53p in RAD53 sml1Δ and in rad53-rep sml1Δ strains after activation of the S phase checkpoint. When wild type cells were treated with HU, Rad53 was hyperphosphorylated, as indicated by an upward shift of the Rad53 band on a western blot ( and C, Lane 2) and by a high intensity band in ISA ( and Lane 2). The rad53-rep mutant exhibited a similar pattern regarding autophosphorylation of Rad53p: extracts from HU treated rad53-rep cells yielded a slower moving Rad53 band in western blots ( and C, Lanes 5 and 6) and a high intensity band of autophosphorylated Rad53p ( and Lane 6) in ISA. Therefore, Rad53 autophosphorylation activity is intact in the rad53-rep mutant during the DNA damage checkpoint. Interestingly, basal levels of Rad53 protein were increased in the rad53-rep mutant compared to wild type. This overexpression phenotype appears to be linked to the separation of function mutation, because the rad53 H426R single mutant, which retains the separation of function phenotype, also expressed elevated levels of Rad53p ( and Lane 7).
We next examined the trans phosphorylation activity of immune complexes containing wild type Rad53p, Rad53-rep p or the kinase dead protein Rad53 K227A D339A. rad53Δ cells were transformed with plasmids expressing HA-tagged wild type RAD53, rad53-rep or rad53 K227A D339A (kinase dead; rad53kd) alleles under control of the GAL1 promoter. Anti-HA antibodies immunoprecipitated HA-tagged Rad53 from untreated transformants and from transformants treated with HU. To examine the trans kinase activity of the immunoprecipitated Rad53 complexes, immunoprecipitates were incubated with γ-32P ATP and histone H1 protein as the kinase substrate. The intensities of histone phosphorylation were quantified by Phosphorimager analysis () and then normalized to levels of immunoprecipitated Rad53 protein, as determined by western blot (). As shown previously in reference Citation50, we found that HU treatment induced a three to eight-fold increase in kinase activities of wild type Rad53, and the same increase was observed with Rad53-rep immune complexes. Furthermore, there were no statistically significant differences between the kinase activities of wild type Rad53 and Rad53-rep immune complexes from untreated cells as well as immune complexes from HU treated cells. We conclude that the kinase activity of Rad53-rep is intact, and upon activation of the DNA replication checkpoint, Rad53-rep kinase activity is activated to levels similar to those of wild type Rad53. These results, coupled with the finding the rad53-rep mutant is resistant to HU, UV and other genotoxic agents (), indicate that Rad53-rep is fully functional in the S-phase and the DNA damage checkpoints.
Rad53-rep protein is defective in binding to ARS1.
During the checkpoint response, Rad53 is associated with arrested replication forks, as shown by chromatin immunoprecipitation experiments, but this association is barely detectable, especially when compared to association levels of other checkpoint proteins such as Tof1p or Mrc1p with paused replication forks.Citation51 Attempts by our lab to detect association of Rad53 with origins of replication in late G1/early S phase have been unsfuccessful (Tecklenburg M and Sclafani RA, unpublished data). Rad53 binds to origin DNA by its kinase domain in the one hybrid assay,Citation46 so we tested whether the replication-defective Rad53-rep protein associated with the ARS1 origin of replication. In addition to Rad53,Citation46 the one hybrid assay has also detected association of DNA replication proteins Orc6 and Dbf4 with ARS1.Citation52,Citation53 In this assay, the reporter gene lacZ is fused downstream of a DNA binding site of the protein of interest. Hybrid proteins fused with transcriptional activation domains (AD) that bind to the target DNA sequence activate transcription of the lacZ reporter gene. For our experiments, the ARS1 origin of replication was fused upstream of the lacZ reporter, and Rad53 or its kinase domain was fused to the B42 transcriptional AD.Citation31,Citation46
The strongest signal was obtained in the X-Gal plate assay with the Rad53 kinase domain fusion (), and reduced signal was seen with the full length wild type RAD53 fusion46 and with the rad53 R70A N107A NVS655-657 AAA fusion (rad53-FHA), a checkpoint-defective, replication-proficient allele with mutations in both the FHA1 and FHA2 domains of RAD53.Citation32,Citation46 Strong signal was also observed with the rad53 K227A D339A kinase dead mutant, indicating that Rad53 kinase activity is not required for association of Rad53 protein with ARS1-lacZ. In contrast, the rad53-rep fusion showed decreased signal compared to the wild type RAD53 fusion. Furthermore, mutating histidine 426 to arginine almost completely eliminates interaction of the Rad53 kinase domain fusion with ARS1-lacZ ( and lower part). Taken together, these data from the one-hybrid assays provide evidence that rad53-rep protein is defective in associating with the ARS1 origin of replication and that histidine 426 of Rad53 is important for interaction of Rad53 with the ARS1 origin of replication.
rad53-rep mutants exhibit a minichromosome loss defect.
Mutations in many genes involved in the initiation of DNA replication such as mcms, orcs and rad53 cause a minichromosome loss phenotype.Citation46,Citation54 The presence of additional ARS elements to the minichromosome increases the probability of establishing an active origin of replication. Therefore, in mutants defective in the initiation of DNA replication, adding extra ARS elements to the circular minichromsome suppresses the loss defect because of mass action.Citation46,Citation55–Citation57 RAD53+ sml1Δ, rad53-rep sml1Δ and rad53Δ sml1Δ strains were transformed with plasmids containing either one ARS or eight ARS elements (). The rad53-rep mutant, like the rad53Δ mutant,Citation46 lost the plasmid containing one ARS at a rate approximately 4 times higher than the loss rate in a wild type strain. The elevated loss rates of rad53Δ and rad53-rep mutants were completely suppressed by additional ARS elements on the plasmid. This phenotype is similar to cdc6, orc2 and mcm mutants defective in the initiation of replication rather than the elongation stage of replication.Citation55–Citation57 We conclude that like rad53Δ mutants, rad53-rep mutants are defective in the initiation of DNA replication. Because Rad53-rep is proficient on the replication and DNA damage checkpoints (–), we also conclude that Rad53's function in the initiation of replication is independent of its role in the DNA damage checkpoint.
Deletion of the major histone H3 H4 gene pair suppresses the synthetic lethality of rad53-rep and cdc7-1.
Rad53 plays an important role in the degradation of excess non-nucleosomal histones.Citation43,Citation44 Deletion of the major copy of the histone H3–H4 gene pair, HHT2-HHF2 (hht2-hhf2Δ) partially suppresses the rad53Δ phenotype regarding chromosome loss, DNA damage sensitivity and slow growth,Citation43 and it also suppresses the synthetic lethality of cdc7-1 and rad53Δ,Citation46 suggesting the function of Rad53 in replication initiation may be related to its function in promoting degradation of excess histones. To determine whether deletion of the HHT2-HHF2 gene pair rescues the synthetic lethality of rad53-rep and cdc7-1, we examined whether a hht2-hhf2Δ URA3-rad53-rep cdc7-1 rad53Δ sml1Δ strain was dependent on a TRP1 RAD53 plasmid for viability (). rad53-rep is synthetically lethal with cdc7-1, thus the HHT2 HHF2+ URA3-rad53-rep cdc7-1 rad53Δ sml1Δ mutant did not grow after counterselection for the TRP1 RAD53 plasmid with 5-FAA, but the hht2-hhf2Δ URA3-rad53-rep cdc7-1 rad53Δ sml1Δ strain was viable on media containing 5-FAA. Therefore, deletion of the major H3/H4 gene pair suppresses synthetic lethality of rad53-rep and cdc7-1, suggesting the role of Rad53 in DNA replication is closely related to chromatin configuration.
rad53-rep mutants are sensitive to histone overexpression.
rad53Δ sml1Δ mutants are sensitive to overexpression of histones,Citation43 and deletion of the major histone H3–H4 gene pair suppresses synthetic lethality of cdc7-1 with rad53ΔCitation46 and with rad53-rep (), suggesting that the role of Rad53 in DNA replication is connected to its role the degradation of excess cellular histones. In order to determine if the sensitivity of the rad53Δ sml1Δ mutant to excess histones is due to a defect in replication function and not to a defect in checkpoint function, we determined whether the checkpoint-proficient, replication defective rad53-rep mutant was sensitive to altered histone concentrations. A rad53Δ sml1Δ strain containing a LEU2 vector or LEU2 plasmid expressing wild type RAD53, rad53-rep or the checkpoint-defective, replication proficient allele rad53-FHACitation32,Citation46 was transformed with a URA3 plasmid expressing the histone H3 and H4 genes HHT2 and HHF2 under control of the GAL1 and GAL10 promoters, respectively (pRM102), or with an empty URA3 vector. Ura+ Leu+ strains were spotted to plates containing either glucose or galactose as the carbon source (). rad53Δ sml1Δ cells transformed with the rad53-rep LEU2 plasmid and pRM102 exhibited growth defects on media containing galactose, thus rad53-rep sml1Δ mutants, like rad53Δ sml1Δ mutants,Citation43 are sensitive to histone overexpression. Conversely, the cells transformed with pRM102 and with the rad53-FHA LEU2 plasmid exhibited wild type levels of growth on both glucose and galactose. Because histone overexpression suppressed the growth of the rad53-rep sml1Δ mutant but not of the checkpoint-defective, replication-proficient rad53-FHA sml1Δ mutant, we conclude that the function of Rad53 in regulating histone levels is related to its function in DNA replication and is independent of Rad53 function in the checkpoint response.
The rad53-rep mutant accumulates excess free histones.
The histone chaperone Asf1 binds the histone H3/H4 heterodimer,Citation58 and excess histones that have not been incorporated into chromatin associate with Asf1p.Citation59 rad53 mutants defective in the degradation of excess non-nucleosomal histones, such as the kinase defective rad53 K227A mutant and the rad53Δ mutant, accumulate high levels of histone H3 associated with histone chaperones including Asf1p.Citation43,Citation44,Citation60 In order to determine if the rad53-rep mutant is defective in the degradation of excess histones, we examined the level of histone H3 that coimmunonprecipitated with the histone chaperone Asf1p (). A C-terminal 13-Myc epitope was integrated downstream of the endogenous ASF1 gene,Citation61 and approximately equal amounts of Asf1-Myc were immunoprecipitated from log-phase cultures of wild type RAD53, rad53Δ and rad53-rep cells. As shown previously, extracts from the rad53Δ mutant exhibited elevated levels of histone H3 coimmunoprecipitating with Asf1 when compared to the wild type strain.Citation44 Approximately 2 times more histone H3 coimmuniprecipated with tagged Asf1 in extracts from the rad53-rep mutant than in extracts from wild type cells. Because rad53-rep mutants accumulate increased levels of histone H3 associated with the Asf1 histone chaperone, we conclude that like the rad53Δ mutant, the rad53-rep mutant is defective in degrading excess, non-nucleosomal histones.
Discussion
The roles of Rad53 in the DNA damage and replication checkpoints and in the essential function of nucleotide regulation have been well-characterized.Citation17,Citation21 Rad53 also has additional functions in replication initiation and in maintenance of histone levels independent of its checkpoint functions.Citation43,Citation45,Citation46 We provide evidence that Rad53's function in replication initiation is genetically separable from its function in the checkpoint response and that Rad53 function in replication is closely related to chromatin structure. The novel rad53-rep and rad53 H426R mutants are proficient in Rad53 checkpoint function, as shown through cell viability and phosphorylation assays, but they are defective in Rad53 DNA replication function, because both alleles are lethal in a cdc7-1 background. Moreover, rad53-rep mutants exhibited a minichromosome loss phenotype that was suppressed by increasing origin dosage, which indicates rad53-rep mutants are defective in the initiation of replication rather than replication elongation.Citation55 A Rad53-rep fusion protein was defective in associating with the ARS1 origin of replication, suggesting that association of Rad53 with origin DNA is important for its function in replication initiation. Reducing gene dosage of histones H3 and H4 suppressed synthetic lethality of rad53-rep and cdc7-1, and we find that sensitivity of rad53 mutants to increased cellular histone levels is due to a defect in Rad53's replication functions, not because of a defect in Rad53's checkpoint functions. Finally the rad53-rep mutant accumulates excess non-nucleosomal histones, providing further evidence that the role of Rad53 in replication initiation is closely related to its role in maintaining cellular histone levels.
Characterization of rad53-rep suggests that both Rad53 kinase activity and association of Rad53 with origins of replication are necessary for Rad53 function during replication initiation. Wild type Rad53 is targeted to origins of replication through its kinase domain,Citation46 and the Rad53-rep protein fusion is defective in interacting with the ARS1 origin of replication. Interestingly, the kinase dead Rad53 protein fusion, which is defective in replication and checkpoint functions,Citation39,Citation46 associates with ARS1 more efficiently than the wild type protein fusion (). Therefore, while association of Rad53 with origins of replication may be important for Rad53's function during replication initiation, another step dependent on Rad53 kinase activity must also occur for normal initiation of DNA replication. We propose that Rad53 positively regulates DNA replication initiation by phosphorylating a specific substrate or substrates at origins of replication.
Rad53 maintains cellular histone levels by phosphorylating excess non-nucleosomal histones, targeting them for proteasomemediated degradation.Citation43,Citation44 Rad53 function in DNA replication initiation is extremely similar to Rad53 histone regulatory function. Both are dependent on Rad53 kinase activity but are independent of Rad53 function in the checkpoint response and in the regulation of nucleotide levels.Citation43,Citation45,Citation46 Deletion of HHT2-HHF2 partially rescues the growth defects of a rad53Δ mutantCitation43 and the synthetic lethality of rad53 mutants with cdc7-1.Citation46 Overexpression of histones H3 and H4 inhibits the growth of the rad53Δ mutant and of the rad53-rep mutant which is specifically defective in DNA replication, not checkpoint function. Furthermore, like the rad53Δ mutant, the rad53-rep mutant accumulates excess chaperone-associated histones that have not been incorporated into chromatin. These findings lead us to two conclusions. First, the DNA replication function of Rad53 but not the checkpoint function of Rad53 is necessary for its role in histone homeostasis. Second, the function of Rad53 in replication initiation is closely linked to cellular histone levels and possibly to chromatin configuration. Therefore, we propose a model in which Rad53 acts as a “nucleosome buffer” at origins of replication to phosphorylate histones that have not been incorporated into chromatin, targeting them for degradation. By degrading excess cellular histones, Rad53 maintains proper chromatin structure at origins of replication that is necessary for efficient initiation of DNA replication.
The chromatin structure surrounding an origin of replication is critical for origin function. Origins of replication consist of a nucleosome-free ORC binding site flanked asymmetrically by precisely positioned nucleosomes, and this specific chromatin structure is essential for origin function and for pre-RC formation.Citation62–Citation64 By maintaining chromatin structure at origins of replication, Rad53 is indirectly promoting formation and/or activation of pre-RCs and subsequent initiation of DNA replication. We hypothesize that because the rad53-rep mutant accumulates non-nucleosomal histones, Rad53-rep protein is defective in phosphorylating excess soluble histones at origins of replication. The accumulation of excess histones in a rad53-rep mutant may lead to alterations in chromatin structure at origins of replication. These alterations would have an inhibitory effect on pre-RC formation and/or activation, and high levels of DDK function would be needed to overcome these effects.
Why is the rad53-rep mutant defective in replication initiation? The rad53-rep mutant expresses increased levels Rad53 protein, even in the absence of the checkpoint response (). Previous works have shown that overexpression of wild type Rad53 and of the Rad53 kinase domain causes a cell cycle delay in the G1/S transition,Citation65 but we propose that the replication defect of the rad53-rep mutant is unrelated to its Rad53p overexpression phenotype. First, rad53-rep mutants enter S phase with wild type kinetics with regards to budding index and to cell size (Holzen TM and Sclafani RA, unpublished data). Second, rad53-rep is a recessive mutation and phenotypes caused by overexpression of a gene tend to be dominant. It is possible that rad53-rep mutants may express elevated levels of Rad53 protein because mutation of histidine 426 to arginine increases the stability of the Rad53 protein. Alternatively, reduced levels of Rad53 activity in a rad53-rep mutant may activate a positive feedback loop, increasing cellular production of Rad53 protein.
A recent study showed that accumulation of free cellular histones caused by mutations in the FACT chromatin modulator or by overexpression of histone genes causes downregulation of G1 cyclin CLN3 and a subsequent delay in G1.Citation60 We also argue that the replication defect of the rad53-rep mutant is unrelated to general cell cycle defects related to an excess of nonnucleosomal histones. First, as mentioned above, the budding index of a rad53-rep mutant released from G1 arrest is similar to that of wild type cells, suggesting that Cln3 activates CDK normally in the rad53-rep mutant. Second, overexpression of either histone pair, H2A–H2B or H3–H4, causes G1 delays in wild type cells,Citation60 but the replication function of RAD53 appears to be specifically related to histone pair H3–H4. Deletion of the histone H3–H4 gene pair HHT2-HHF2 suppresses synthetic lethality of cdc7-1 with rad53Δ,Citation46 but deletion of the H2A–H2B gene pair HTA2-HTB2 has no effect on rad53Δ-cdc7-1 synthetic lethality (Tecklenburg M and Sclafani RA, unpublished data).
One possible explanation for the replication defect of rad53-rep is that mutation of histidine 426 to arginine causes decreased affinity of Rad53 kinase for its histone substrate. Comparison of the sequence of S. cerevisiae RAD53 to the sequence and structure of its human homolog CHK2,Citation66 and other eukaryotic protein kinases,Citation67 places histidine 426 in subdomain X of the RAD53 kinase domain.Citation68 Subdomain X is a poorly conserved subdomain in the protein kinase superfamily, and it may play a role in kinase-substrate recognition.Citation69,Citation70 Because Rad53 kinase activity is low in the absence of checkpoint activation ( and reviewed in ref. Citation49), we expect that the substrate of Rad53 kinase during replication initiation has high affinity for the Rad53 kinase domain. According to our proposed model, a RAD53 mutation that decreases its interactions with non-nucleosomal histones would likely cause accumulation of excess histones, chromatin structure specifically at origins of replication would be altered, and DNA replication could not occur when CDC7 function is compromised. It is possible that altered origin chromatin structure may lead to decreased interaction of Rad53 protein with origins of replication, as seen in the rad53-rep mutant. However, the one hybrid data presented here do not support this model. All one hybrid experiments were performed in a rad53Δ strain background. Both rad53Δ and rad53 K227A D339A kinase dead mutants accumulate excess histones,Citation43,Citation44,Citation60 thus if excess histones inhibit interaction of Rad53 with ARS1, we would expect to see decreased interaction of the Rad53 K227A D339A fusion protein with origins of replication. However, we detect strong interaction of the Rad53 K227A D339A fusion protein with ARS1, suggesting that excess histone levels do not affect interaction of Rad53 with origins of replication.
In another possible explanation, the replication defect of rad53-rep mutants may directly result from decreased interaction of Rad53-rep with origins of replication. Therefore, the excess histones that accumulate in the rad53-rep mutant result from decreased levels of Rad53 activity at origins of replication and are not causing the replication defect in the rad53-rep mutant per se. The Rad53-rep fusion protein is defective in interacting with ARS1 in the one hybrid assay, suggesting that interaction of Rad53 with origin DNA is important for its function in replication. Mutation of histidine 426 to arginine may inhibit interaction of Rad53 protein with origins of replication. If Rad53 specifically acts at origins of replication to maintain the chromatin structure of the origin, mutations that negatively affect interaction of Rad53 with origin DNA would cause altered chromatin structure at the origins of replication. These types of rad53 mutants would then inhibit initiation of DNA replication, because chromatin structure is central to origin function.Citation62–Citation64
Finally, it is possible that both association of Rad53 with origins and Rad53-mediated degradation of histones are central to Rad53's function during replication initiation and that histidine 426 of Rad53 is important for both of these functions. Mutation of histidine 426 as in rad53-rep and rad53-H426R mutants causes decreased interaction of Rad53p with origins of replication and accumulation of excess histones. Loss of both of these activities causes the replication defects such as the plasmid loss phenotype of the rad53-rep mutant. Furthermore, when Cdc7 activity is compromised, defects in both of these Rad53 functions cause a replication initiation defect which is lethal to the cell.
Rad53 phosphorylates tyrosine residue 99 of histone H3, and the ubiquitin conguating enzymes Ubc4 and Ubc5 and the ubiquitin ligase Tom1 have been identified as E2 and E3 ligases, respectively, involved in the degradation of excess soluble histones.Citation44 We have not detected a genetic interaction between RAD53 or CDC7 with TOM1 (Holzen TM and Sclafani RA, unpublished data), but it is possible that other E2 or E3 ligases can substitute for Tom1 in degradation of excess histones.Citation71,Citation72 As more histone variants and more ubiquitin ligases involved in histone degradation are identified, it will be interesting to examine whether mutations in any of these genes also affect replication initiation.
The work presented provides evidence that Rad53 plays an important but nonessential role in the initiation of DNA replication that is genetically separable from its role in the DNA damage and S phase checkpoints. Genetic interaction between the replication-defective rad53-rep allele with the histone H3–H4 gene pair HHT2-HHF2 provides strong support for a model in which Rad53 function in replication initiation is closely linked to chromatin structure. Previous studies have emphasized the importance of chromatin structure in the initiation of DNA replication, and origin function is dependent on the chromatin structure of the origin and the adjacent regions of DNA.Citation62–Citation64 Genome-wide studies have also suggested that RAD53 may play a part in the maintenance of chromatin structure,Citation73 and deletion of HIR2 or HIR3, genes encoding co-repressors of histone gene transcription, is synthetically sick with temperature sensitive alleles of CDC7 or the replication protein CDC45.Citation74,Citation75 Finally, the separation of function allele rad53-rep will be a very useful tool for precisely defining the role of Rad53 in the initiation of replication.
Materials and Methods
Yeast strains, media and plasmids.
Yeast strains were grown in yeast extract/peptone/dextrose (YEPD) with 2% glucose or in synthetic defined (SD) minimal media supplemented with appropriate amino acids and 2% glucose. All strains are congenic with A364a,Citation76 except as specified (). Standard genetic methods were used for strain construction and tetrad analysis, and transformation of yeast strains was performed by the lithium acetate method.Citation77
All plasmids used in this study are listed in . pTH4A (pRS315-RAD53) was constructed by creating a unique AvrII site immediately downstream of the RAD53 ORF in pPD361 (pRS315-RAD53) with the Quikchange kit (Stratagene) and primers oilTH42 (5′-GAG AAT TGG CAA TTT TCG TAA CCT AGG ACA AAT ACC CAT AGA AAA TG-3′) and oliTH43 (5′-CAT TTT CTA TGG GTA TTT GTC CTA GGT TAC GAA AAT TGC AAA TTC TC-3′). To create pTH15 (pRS315 rad53 H426R), pTH4A (pRS315-RAD53) was mutagenized with the Quikchange kit using primers oliTH59 (5′-ATT GGA AGA GGC TCA TAT CGT GAA GGG CCC CTC-3′) and oliTH60 (5′-GAG GGG CCC TTC ACG ATA TGA GCC TCT TCC AAT-3′).
For pTH16 (pRS306-RAD53) and pTH17 (pRS306-rad53-rep), the 5.5 kb ClaI fragment of pTH4A and pTH13, repectively, was ligated into the ClaI site of the pRS306 polylinker. For pTH18 (pRS306-rad53RN/NVS-AA/AAA), the 3.5 kb EcoRI fragment containing rad53RN/NVS-AA/AAA of pTH14 (pRS315-rad53-FHA; gift from D. Stern) was ligated into the EcoRI site of the polylinker of pRS306. pTH19 (pRS306-rad53 H426R) was constructed by removing the 2.5 kb NotI-BspEI fragment containing the first 1.3 kb of RAD53 from pRS306-RAD53 and ligating a 2.5 kb NotI-BspEI fragment from pTH15 (pRS315-rad53 H426R) in the respective sites. To integrate the pRS306-RAD53 plasmids at the URA3 locus, pRS306-rad53 plasmids were digested with StuI and transformed into ura3 yeast strains. All integrations were confirmed by Southern Blots of BglII and BspEI digested genomic DNA.
pBJ371 (ARS CEN LEU2 mcm4Δ74-174) was obtained from B. Stillman. A 4.8 kb SacI-HindIII fragment of pBJ371 was cloned into pRS305 to produce pRAS753 (PRS305-mcm4Δ74-174). A 1.7 kb SalI fragment of pRAS753 was deleted to produce pRAS755 (pRS305-mcm4Δ74-174 3′“Δ), which has the 3′ end of the mcm4 gene deleted. TMH443 was transformed with MluI-digested pRAS755 to create RSY1363, which contains a partial duplication of the mcm4 gene, with only the mcm4Δ74-174 gene being active.
Plasmids containing B42AD-RAD53 (pRAD53) and B42AD-rad53 (G177-N599) fusions used in the one-hybrid and immunoprecipitation/H1 kinase assays have been described previously in reference Citation31. pTH25 (B42AD-rad53-repTRP1) was constructed by removing the 1.7 kb Eco91I and SwaI fragment of wild type RAD53 from pRAD53 and ligating the 1.7 kb Eco91I-SwaI fragment of rad53-rep from pTH13 at the respective sites. pTH26 (B42AD-rad53-FHA TRP1) was constructed by digesting pTH18 with AflII, and the 2 kb fragment was cloned into the corresponding sites of pRAD53 digested with AflII. pTH27 (B42AD-rad53-K227A D339A TRP1) was constructed by amplifying a 2.2 kb fragment of rad53 K227A D339A with primers oliTH66 (5′-AAT CCA CGC AGG CTA CTC AAA GGT-3′) and oliTH67 (5′-AGG GTT GAT TTG CAC GTT GAG CTG-3′) with pRS306-PGAL rad53 K227A D339A (gift from O. Aparicio) as a template. pRAD53 was digested with AflII, and the DNA fragments were resolved by agarose gel electrophoresis to separate of a 2 kb fragment RAD53 (nt 167–2,150) from the plasmid. The yeast strain TMH327 was co-transformed with the 7 kb fragment representing the cut plasmid and the rad53 K227A D339A PCR product, and the B42AD-rad53 K227A D339A plasmid was formed by homologous recombination in the yeast. pTH35 (TRP1 B42AD-RAD53 G177-N599, H426R) was created by mutagenizing pKIN (TRP1 B42AD-RAD53 G177-N599) with the Quikchange kit and primers oilTH59 and oliTH60.
A C-terminal 13-Myc epitope tag was integrated at the endogenous copy of ASF1,Citation61 using a PCR product from primers yASF1 mid (5′-GAA TAA TGA GTA CGA TGA AG-3′) and ASF1ScrRev (5′-CTA TAC ATT GAT GTG ACG AC-3′) and a genomic prep of yeast strain BKD187, a W303 strain containing the tagged ASF1 construct, as a template. Primers and template were gifts from B. Dennehey.
RAD53 mutagenesis.
Random mutagenesis of the kinase domain of RAD53 was performed by error-prone PCR. Primers oliTH10 (5′-TAA GCA GTG CCT CGA GCA GAA CAA-3′) and oliTH11 (5′-GCT GCG ACA ACG ATT GTG ATA AGG-3′) amplified nucleotides 453-1,488 of the RAD53 ORF under the following conditions: 1x PCR Buffer (20 mM Tris-HCl, pH 8.4; 50 mM KCl; Invitrogen); 3.5 mM MgCl2; 50 µM MnCl2; 0.2 mM dNTPs; 0.2 mM of each primer; 1 ng template DNA (pTH4A); and 2 units Taq DNA polymerase (Invitrogen). PCR was performed in a Biometra T3 Thermocycler with an initial denaturation at 95°C, followed by 30 cycles of 30 sec at 95°C, 30 sec at 55°C and 1 min at 72°C. The following conditions yielded a nucleotide base substitution of 1.34 × 10-2 substitutions/nucleotide. Independent reaction mixtures were combined and purified using a QIAquick Spin Column (QIAGEN). pTH4A was digested with MscI and NdeI, and the DNA fragments from the reaction were resolved by agarose gel electrophoresis to remove a 863 bp fragment of RAD53 representing the kinase domain (nt 562–1,425 of the RAD53 ORF) from the 10.5 kb fragment of linearized pTH4A. The 10.5 kb plasmid DNA fragment was purified, and it was co-transformed with purified PCR product in an approximately 1:1 molar ratio into yeast strain TMH38. Transformants were selected on SD plates lacking leucine incubated at 22°C, and colonies on these plates were replica plated to YEPD plates containing 120 mM hydroxyurea (HU) to select for checkpoint-proficent transformants. HUR colonies were then spotted to SC plates lacking leucine, to YEPD + HU, and to 5-FOA plates to eliminate pPD1 (pARS-CEN-URA3-CDC7) and incubated at 22°C. Plasmid DNA from Leu+ HUR cells that did not grow on 5-FOA were isolated from the yeast cells with the RPM kit (MP Biomedicals, LLC) and transformed into E. coli strains by electroporation.
Plasmid shuffle assays.
For URA3 plasmid loss, cells were spotted to plates containing 1.5 g/L 5-fluoroorotic acid (5-FOA). For TRP1 plasmid loss, cells were spotted to plates containing 0.75 g/L 5-fluoroanthranilic acid (5-FAA).
DNA damage survival assays.
In the HU-resistance assays, cultures in mid-log phase (2 × 107 cells/ml) were transferred to YEPD + 200 mM HU, and grown at 30°C. Aliquots were removed at designated time points, plated to YEPD, and grown at 30°C for 2–3 days, after which colonies were counted. UV-resistance assays were performed as described previously in reference Citation78. In the MMS and zeocin survival assays, mid-log cultures of cells were diluted to an initial concentration of 1 á 107 cells/ml, 10-fold serial dilutions were spotted to YEPD plates as a control, YEPD plates + 0.05% MMS (Sigma) and YEPD plates + 15 mg/ml Zeocin (Invitrogen) and plates were incubated at 30°C for two days.
Western blotting.
Yeast protein extracts were prepared from TCA-treated cells79 and protein extracts were resolved by 10% SDS-PAGE and transferred to nitrocellulose or PVDF membrane. Membranes were probed with anti-Rad53 antibody from rabbit (gift from D. Stern) at a 1:2,000 dilution or with anti-Rad53 antibody from rabbit (gift from N. Lowndes) at a 1:10,000 dilution. Rabbit anti-glucose 6 phosphate dehydrogenase (Sigma) at a 1:100,000 dilution.
To analyze coimmunoprecipitation of histone H3 with Asf1-Myc, membranes were probed with mouse anti-Myc antibody (9E10; Babco) at a concentration of 0.2 µg/ml or with rabbit histone H3 antibody (Abcam) at a concentration of 0.3 µg/ml.
Secondary horseradish peroxidase-conjugate goat anti-rabbit antibody or goat-anti mouse antibody (BioRad) was used at a 1:10,000 dilution. A 1:20,000 dilution of goat anti-Rabbit IR Dye 680 (LI-COR) was used for Rad53 immunoblots in the IP Kinase Assay. 1:15,000 dilutionsof goat anti-Rabbit IR Dye 680 and goat anti-Mouse IR Dye 800 were used for Asf1-Myc and Histone H3 immunoblots in Asf1-Myc immunoprecipitations. Immunoblots were visualized with an ECL chemiluminescence kit (Thermo Scientific) or visualized with the Odyssey Infrared Imaging System (LI-COR) and quantified with Odyssey v. 2.1 software (LI-COR).
Kinase assays.
For the in situ autophosphorylation assay, mid-log phase cultures of TMH115 (rad53-rep), TMH116 (rad53Δ) and TMH122 (RAD53) were incubated in YEPD+200 mM hydroxyurea (Sigma) for three hours or left untreated. Yeast protein extracts were then prepared by TCA extraction,Citation79 and the in situ autophosphorylation assay was performed as described previously in reference Citation49.
Rad53 immune complex kinase assays.
Rad53 immunoprecipitation kinase assays were performed as described previously in reference Citation50 and Citation65. rad53Δ strains expressing plasmid-borne HA-tagged alleles of RAD53 under control of the GAL1 promoter (pRAD53, pTH25 or pTH27) were grown to saturation in -Trp+2% Raffinose. Overexpression of RAD53 was induced by resuspending cells to a final concentration of 2 × 107 cells/ml in YEP+2% Raffinose + 2% Galactose. For HU treated cells, HU was added to a final concentration of 200 mM and cultures were incubated overnight at 30°C. Untreated cultures were incubated in YEP+2% Raffinose + 2% Galactose for three hours to induce overexpression of Rad53p. Cells were lysed with glass beads in HEPES Lysis buffer (25 mM HEPES pH 7.5; 10% glycerol; 0.1% Triton X-100; 1 mM EDTA; 10 mM Sodium Fluoride; 20 mM β-glycerophosphate; 5 mM sodium vanadate; and protease inhibitor cocktail (Roche)). 3 mg protein was incubated overnight with 25 µg mouse anti-HA antibody (Roche) at 4°C. 40 µl Protein G Plus/Protein A Agarose beads (Calbiochem) were added to immunoprecipitations and incubated at 4°C for one hour. Beads were washed in Kinase Wash Buffer (25 mM HEPES, pH 7.5; 10% glycerol; 0.1% Triton X-100; 1 mM EDTA) three times. IP Kinase assays were performed on ice for seven minutes in 25 µl IP Kinase Reaction Buffer (25 mM HEPES, pH 7.5; 1 mM MnCl2; 1 mM MgCl2; 10 µCi γ-32P ATP; 1 µM ATP) with 4 µg Histone H1 as substrate per reaction. An equal amount of boiling 2x SDS-PAGE loading buffer was added to each sample and boiled. Reactions were resolved on a 10% SDS-PAGE gel. The portion of the gel below the 55 kDa membrane was stained with Coomassie blue, dried and exposed to a Phosphor Screen (Molecular Dynamics), scanned and phosphorylation of Histone H1 was quantified using ImageQuant 5.2 software (Molecular Dynamics). The portion of the gel above approximately 55 kDa was transferred to nitrocellulose membrane (Whatman) and immunoblotted for Rad53.
One-hybrid assay.
X-Gal plate assays were performed as previously described in references Citation46 and Citation80.
Minichromosome loss assays.
Mitotic loss rates were calculatedCitation81 for strains transformed with plasmids containing one ARS (pDK243) or eight ARS elements (pDK368-7). All loss rates are percentages per generation and were determined in triplicate for each experiment.
Histone overexpression assays.
TMH128 was co-tranformed with pRS316 (URA3 vector) or pRM102 (pURA3 PGAL1 HHT2 PGAL10 HHF2,Citation82) and pRS315 (LEU2 vector); pPD361 (pLEU2 RAD53); pTH13 (pLEU2 rad53-rep); or pTH14 (pLEU2 rad53-FHA). Ura+ Leu+ transformants were grown for three days at 30°C in SD+2% raffinose liquid media lacking leucine and uracil to a concentration of approximately 2 × 107 cells/ml. Synthesis of histones H3 and H4 was induced by adding galactose to a final concentration of 2% and cultures were incubated at 30°C for three hours. 10-fold serial dilutions of cultures were spotted to SD plates lacking leucine and uracil, with either 2% glucose or galactose as the carbon source. Plates were incubated for four days at 30°C.
Immunoprecipitation of Asf1-13Myc.
Myc-tagged Asf1p was immunoprecipated as described previously in reference Citation43. Cells from 1 liter mid-log phase cultures (≈2 × 107 cells/ml) were harvested, washed with ddH2O, resuspended in 5 ml lysis buffer (20 mM HEPES-KOH, pH 7.5; 110 mM potassium acetate; 10% glycerol; 0.1% Tween-20; 1 mM sodium vanadate; 50 mM sodium fluoride; 50 mM sodium β-glycerophosphate; 10 mM sodium butyrate; 10 mM β-mercaptoethanol and 1x Roche protease inhibitors) and frozen in liquid nitrogen. Whole cell extracts were prepared by grinding frozen cell pellets in liquid nitrogen with a SPEX SamplePrep 6870 Freezer Mill. 10 ml additional lysis buffer were added to the ground cell pellets and lysates were clarified by centrifuging twice at 13,000x g for 10 minutes. Protein concentrations of the extracts were determined with the BCA Protein Assay kit (Pierce). 5 mg of protein extract was mixed with 40 µl c-Myc (9E10) antibody agarose beads (Santa Cruz Biotech) to immunoprecipitate Asf1-13Myc or with 40 µl IgG Sepharose beads blocked in Lysis Buffer + 0.1% BSA as a no antibody control, and samples were incubated with end-over-end rotation overnight at 4°C. Beads were collected by centrifugation at 2,500x g, the supernatants were saved and the beads were washed twice with 1 ml wash buffer (20 mM HEPES-KOH, pH 7.5; 110 mM potassium acetate; 10% glycerol; 0.1% Tween-20) and once with 1 ml PBS. 40 µl 2x sample buffer was added to the beads, boiled and separated on SDS-15% polyacrylamide gels, along with 50 µl (5%) of whole cell extracts and of immunoprecipitation supernatants. The gel was blotted to nitrocellulose membrane (Whatman), which was immunoblotted for Asf1-13Myc and for histone H3.
Figures and Tables
Figure 1 The recessive separation of function allele rad53-rep confers resistance to HU and is synthetically lethal with cdc7-1. (A) A 1:10 dilution series of saturated cultures of TMH38 leu2 ura3 cdc7-1 rad53Δ sml1Δ pCDC7 URA3 transformed with indicated LEU2 plasmids were spotted to -Leu dropout media, YEPD+120 mM HU and 5-FOA plates and grown at 22°C for three days: wild type RAD53 (pTH4A), empty LEU2 vector (pRS315), rad53-rep (pTH13) and rad53 H426R (pTH15). Only cells with pRAD53 LEU2 (pTH4A) can lose pCDC7 URA3 (pPD1) and grow on the 5-FOA plate. (B) A 1:10 dilution series of saturated cultures of the following trp1 sml1Δ cdc7-1 pRAD53 TRP1 derivatives was spotted to YEPD and 5-FAA plates and grown at 22°C for three days: URA3-RAD53 rad53Δ (TMH154), URA3 rad53Δ (TMH150), URA3-rad53-rep rad53Δ (TMH152), URA3-rad53-rep RAD53 (TMH437), ura3 RAD53 (TMH404). (C) A 1:10 dilution series of saturated cultures of the following trp1 rad53Δ sml1Δ cdc7-1 pRAD53 TRP1 derivatives was spotted to YEPD and FAA plates and grown at 30°C for three days: URA3-RAD53 mcm5-bob1 (TMH148), rad53Δ mcm5-bob1 (TMH146), URA3-rad53-rep mcm5-bob1 (TMH147). In (B and C), cells lacking a chromosomal copy of RAD53 are dependent on pRAD53 TRP1 (pPD83) for growth and cannot grow on the 5-FAA plate.
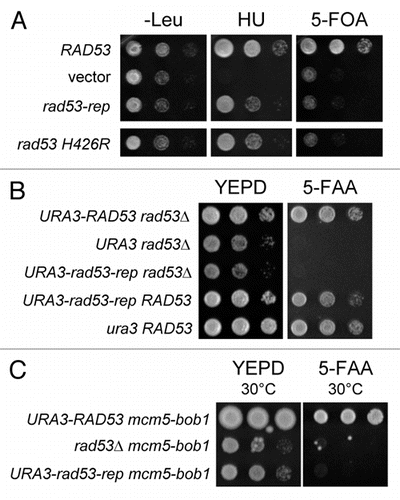
Figure 2 The DNA damage checkpoints are intact in the rad53-rep mutant. ◯ RAD53 sml1Δ (TMH122, TMH337); △ rad53-rep sml1Δ (TMH115, TMH358); □ rad53 H426R sml1Δ (TMH453); × (: rad53Δ sml1Δ (TMH116, TMH351). (A) Logarithmically growing cultures were incubated in YEPD + 0.2 M HU for the indicated times. Cell viability was measured as colony formation on YEPD after incubation in HU. (B) Equal numbers of cells from logarithmically growing cultures were plated onto YEPD and irradiated with increasing doses of ultraviolet light. Colonies were counted after two days of incubation at 30°C in the dark to determine viability. (C) A 1:10 dilution series of logarithmically growing cells was spotted to YEPD, YEPD + 0.05% MMS and YEPD + 15 mg/ml zeocin and plates were incubated at 30°C for two days.
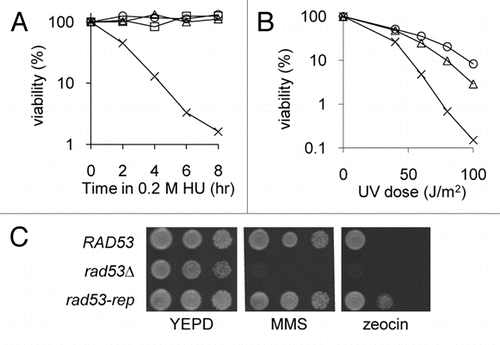
Figure 3 Rad53 protein is autophosphorylated in the rad53-rep mutant after treatment with HU. Log phase cultures of RAD53 (TMH122; WT), rad53Δ (TMH116; Δ), rad53-rep (TMH115; rep) and rad53-H426R (TMH 453; H426R) strains were treated in 0.2 M HU for three hours. Protein extracts were prepared from log phase (L) and HU-treated (H) strains and analyzed by western blotting (A and C) using Rad53 antibodies and glucose-6-phosphate dehydrogenase (G6PD) antibodies as a loading control or by the in situ autophosphorylation assay (B).
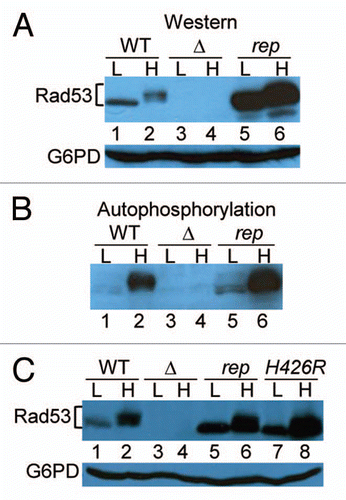
Figure 4 trans-phosphorylation activity of Rad53-rep immune complexes. RSY1046 was transformed with the designated plasmids, and HA-tagged wild type Rad53 (pRAD53), rad53-rep (pTH25) and rad53 K227A D339A (rad53-kd; pTH27) were immunoprecipitated with antibody against HA (+) or mock immunoprecipitated (-) from lysates of untreated cells (UN) and from cells treated with HU (HU). Immune complexes were incubated on ice with Histone H1 in the presence of γ-32P ATP. (A) Rad53 western blot of immunoprecipitations. (B) Autoradiogram of Histone H1 phosphorylated by Rad53 immune complexes.
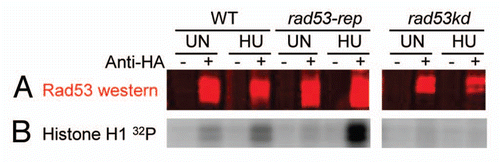
Figure 5 Rad53-rep is defective in binding ARS1 due to the H426R mutation. Strain RSY1064 with an ARS-lacZ reporter was transformed with plasmids carrying different Rad53-AD fusions. Transformants were assayed for β-galactosidase activity using the X-Gal filter assay. Plasmids used in this assay are as follows: wild type (pRAD53), rad53-rep (pTH25), rad53-FHA (pTH26), rad53 K227A D339A (pTH27), vector (pJG4-6), kinase domain (Rad53 G177-N599, pKIN), kinase domain H426R (pTH35).

Figure 6 The rad53-rep mutant exhibits a minichromosome loss phenotype. RAD53 (TMH122), rad53-rep (TMH115) and rad53Δ (TMH116) sml1Δ strains were transformed with plasmids containing either one ARS (pDK243) or eight ARS elements (pDK368-7). Mitotic loss rates were calculated by fluctuation tests. The data represent mean values calculated from three independent experiments.
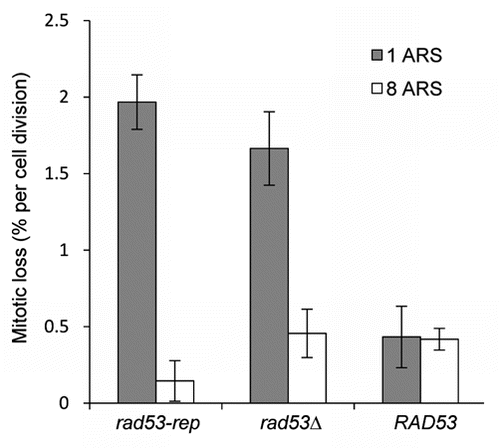
Figure 7 Deletion of the major histone H3 H4 gene pair suppresses synthetic lethality of rad53-rep and cdc7-1. cdc7-1 rad53Δ sml1Δ (pRAD53 TRP1) strains with RAD53 (TMH154), rad53-rep (TMH152) or empty pRS306 integration vector (TMH150) at the URA3 locus were transformed with a hht2-hhf2Δ::HIS3 deletion cassette to generate strains TMH159, TMH160 and TMH164 respectively. A 1:10 dilution series of saturated cultures of the strains were spotted to YEPD and 5-FAA plates and grown at 22°C for three days. Cells with a chromosomal copy of wild type RAD53 or the hht2-hhf2Δ::HIS3 deletion can lose the pRAD53-TRP1 plasmid and grow on the 5-FAA plate.
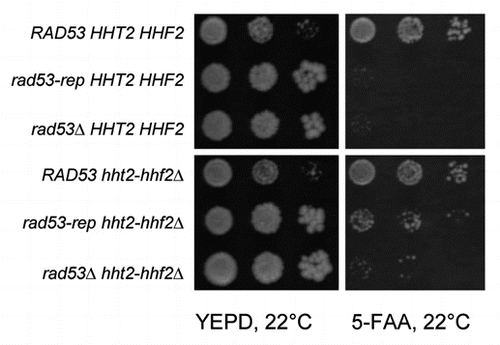
Figure 8 The rad53-rep mutant is sensitive to histone overexpression. A ura3 leu2 rad53Δ sml1Δ strain (TMH128) was transformed with the designated LEU2 RAD53 plasmids and a URA3 vector containing histone H3 H4 genes HHT2 and HHF2 under control of the GAL1 and GAL10 promoters (pRM102) or an empty URA3 vector (pRS316). Expression of histones H3 and H4 was induced by adding galactose to liquid cultures of the transformants. A 1:10 dilution series of the cultures was spotted to minimal plates lacking uracil and leucine, with either glucose or galactose as the sole carbon source, and the plates were incubated at 30°C for three days. RAD53 LEU2 plasmids used: RAD53 (pTH4A), rad53Δ (pRS315), rad53-rep (pTH13) and rad53-FHA (pTH14).
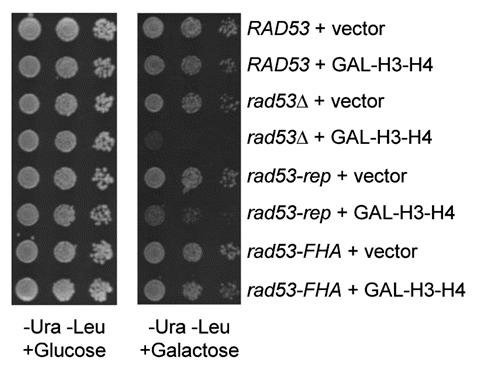
Figure 9 The rad53-rep mutant accumulates excess endogenous histones bound to histone chaperone Asf1. Myc-tagged Asf1 was immunoprecipitated from lysates from wild type (RAD53; TMH483), rad53-rep (TMH484), rad53Δ (TMH488) strains, and from a rad53Δ strain expressing untagged Asf1 (No tag; TMH90). Immunoprecipitations were analyzed by western blotting with antibodies against Myc and histone H3. W, S and P refer to whole cell extracts, supernatant and pellet fractions, respectively. For each tagged strain, the intensity of the band of co-immunoprecipitated histone H3 was normalized to the intensity of the band of immunoprecipitated Asf1-Myc.
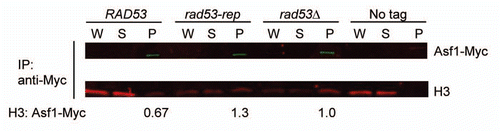
Table 1 Yeast strains used in this study
Table 2 Plasmids used in this study
Acknowledgements
We thank members of the Sclafani lab for helpful discussions, Oscar Aparicio, David Stern and Bruce Stillman for plasmids, Noel Lowndes and David Stern for Rad53 antibodies and B. Dennehey of J. Tyler's laboratory for primer sequences and genomic DNA templates. The DNA samples were sequenced by the University of Colorado Cancer Center DNA Squencing and Analysis Core Facility. This work was supported by a US Public Health Service grant GM35078 awarded to R.A.S.
References
- Takeda DY, Dutta A. DNA replication and progression through S phase. Oncogene 2005; 24:2827 - 2843
- Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet 2007; 41:237 - 280
- Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JF. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature 1996; 379:180 - 182
- Tanaka S, Diffley JF. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat Cell Biol 2002; 4:198 - 207
- Labib K, Tercero JA, Diffley JF. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 2000; 288:1643 - 1647
- Bochman ML, Schwacha A. The Mcm2-7 complex has in vitro helicase activity. Mol Cell 2008; 31:287 - 293
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, et al. Replication dynamics of the yeast genome. Science 2001; 294:115 - 121
- Tanaka S, Umemori T, Hirai K, Muramatsu S, Kamimura Y, Araki H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007; 445:328 - 332
- Zegerman P, Diffley JF. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 2007; 445:281 - 285
- Sclafani RA. Cdc7p-Dbf4p becomes famous in the cell cycle. J Cell Sci 2000; 113:2111 - 2117
- Murray AW. Recycling the cell cycle: cyclins revisited. Cell 2004; 116:221 - 234
- Hardy CF, Dryga O, Seematter S, Pahl PM, Sclafani RA. mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc Natl Acad Sci USA 1997; 94:3151 - 3155
- Sheu YJ, Stillman B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010; 463:113 - 117
- Fletcher RJ, Chen XS. Biochemical activities of the BOB1 mutant in Methanobacterium thermoautotrophicum MCM. Biochemistry 2006; 45:462 - 467
- Hoang ML, Leon RP, Pessoa-Brandao L, Hunt S, Raghuraman MK, Fangman WL, et al. Structural changes in Mcm5 protein bypass Cdc7-Dbf4 function and reduce replication origin efficiency in Saccharomyces cerevisiae. Mol Cell Biol 2007; 27:7594 - 7602
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet 2002; 36:617 - 656
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9:297 - 308
- Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989; 246:629 - 634
- Zhou Z, Elledge SJ. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell 1993; 75:1119 - 11127
- Bashkirov VI, Bashkirova EV, Haghnazari E, Heyer WD. Direct kinase-to-kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol Cell Biol 2003; 23:1441 - 1452
- Segurado M, Tercero JA. The S-phase checkpoint: Targeting the replication fork. Biol Cell 2009; 101:617 - 627
- Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. Replication in hydroxyurea: it's a matter of time. Mol Cell Biol 2007; 27:6396 - 6406
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, et al. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001; 412:557 - 561
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002; 297:599 - 602
- Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001; 412:553 - 557
- Segurado M, Diffley JF. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev 2008; 22:1816 - 1827
- Hofmann K, Bucher P. The FHA domain: A putative nuclear signalling domain found in protein kinases and transcription factors. Trends Biochem Sci 1995; 20:347 - 349
- Durocher D, Jackson SP. The FHA domain. FEBS Lett 2002; 513:58 - 66
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, et al. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 2001; 3:958 - 965
- Smolka MB, Chen SH, Maddox PS, Enserink JM, Albuquerque CP, Wei XX, et al. An FHA domainmediated protein interaction network of Rad53 reveals its role in polarized cell growth. J Cell Biol 2006; 175:743 - 753
- Duncker BP, Shimada K, Tsai-Pflugfelder M, Pasero P, Gasser SM. An N-terminal domain of Dbf4p mediates interaction with both origin recognition complex (ORC) and Rad53p and can deregulate late origin firing. Proc Natl Acad Sci USA 2002; 99:16087 - 16092
- Schwartz MF, Lee SJ, Duong JK, Eminaga S, Stern DF. FHA domain-mediated DNA checkpoint regulation of Rad53. Cell Cycle 2003; 2:384 - 396
- Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 1998; 281:272 - 274
- Chen SH, Zhou H. Reconstitution of Rad53 activation by Mec1 through adaptor protein Mrc1. J Biol Chem 2009; 284:18593 - 18604
- Jia-Lin Ma N, Stern DF. Regulation of the Rad53 protein kinase in signal amplification by oligomer assembly and disassembly. Cell Cycle 2008; 7:808 - 817
- Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J 2001; 20:3544 - 3553
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 2003; 112:391 - 401
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev 1994; 8:2401 - 2415
- Fay DS, Sun Z, Stern DF. Mutations in SPK1/RAD53 that specifically abolish checkpoint but not growthrelated functions. Curr Genet 1997; 31:97 - 105
- Pike BL, Yongkiettrakul S, Tsai MD, Heierhorst J. Diverse but overlapping functions of the two forkheadassociated (FHA) domains in Rad53 checkpoint kinase activation. J Biol Chem 2003; 278:30421 - 30424
- Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 1998; 12:2956 - 2970
- Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 1998; 2:329 - 340
- Gunjan A, Verreault A. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 2003; 115:537 - 549
- Singh RK, Kabbaj MH, Paik J, Gunjan A. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 2009; 11:925 - 933
- Dohrmann PR, Oshiro G, Tecklenburg M, Sclafani RA. RAD53 regulates DBF4 independently of checkpoint function in Saccharomyces cerevisiae. Genetics 1999; 151:965 - 977
- Dohrmann PR, Sclafani RA. Novel role for checkpoint Rad53 protein kinase in the initiation of chromosomal DNA replication in Saccharomyces cerevisiae. Genetics 2006; 174:87 - 99
- Zheng P, Fay DS, Burton J, Xiao H, Pinkham JL, Stern DF. SPK1 is an essential S-phase-specific gene of Saccharomyces cerevisiae that encodes a nuclear serine/threonine/tyrosine kinase. Mol Cell Biol 1993; 13:5829 - 5842
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 2003; 17:1755 - 1767
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, et al. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 1999; 18:6561 - 6572
- Lee SJ, Schwartz MF, Duong JK, Stern DF. Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol Cell Biol 2003; 23:6300 - 6314
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, et al. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003; 424:1078 - 1083
- Li JJ, Herskowitz I. Isolation of ORC6, a component of the yeast origin recognition complex by a one-hybrid system. Science 1993; 262:1870 - 1874
- Dowell SJ, Romanowski P, Diffley JF. Interaction of Dbf4, the Cdc7 protein kinase regulatory subunit, with yeast replication origins in vivo. Science 1994; 265:1243 - 1246
- Maine GT, Sinha P, Tye BK. Mutants of S. cerevisiae defective in the maintenance of minichromosomes. Genetics 1984; 106:365 - 385
- Hogan E, Koshland D. Addition of extra origins of replication to a minichromosome suppresses its mitotic loss in cdc6 and cdc14 mutants of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1992; 89:3098 - 3102
- Leon RP, Tecklenburg M, Sclafani RA. Functional conservation of beta-hairpin DNA binding domains in the Mcm protein of Methanobacterium thermoautotrophicum and the Mcm5 protein of Saccharomyces cerevisiae. Genetics 2008; 179:1757 - 1768
- Loo S, Fox CA, Rine J, Kobayashi R, Stillman B, Bell S. The origin recognition complex in silencing, cell cycle progression and DNA replication. Mol Biol Cell 1995; 6:741 - 756
- English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. Structural basis for the histone chaperone activity of Asf1. Cell 2006; 127:495 - 508
- Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell 2005; 17:301 - 311
- Morillo-Huesca M, Maya D, Munoz-Centeno MC, Singh RK, Oreal V, Reddy GU, et al. FACT prevents the accumulation of free histones evicted from transcribed chromatin and a subsequent cell cycle delay in G1. PLoS Genet 2010; 6:1000964
- Tamburini BA, Carson JJ, Adkins MW, Tyler JK. Functional conservation and specialization among eukaryotic anti-silencing function 1 histone chaperones. Eukaryot Cell 2005; 4:1583 - 1590
- Simpson RT. Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature 1990; 343:387 - 389
- Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. Conserved nucleosome positioning defines replication origins. Genes Dev 2010; 24:748 - 753
- Lipford JR, Bell SP. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell 2001; 7:21 - 30
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev 1996; 10:395 - 406
- Oliver AW, Paul A, Boxall KJ, Barrie SE, Aherne GW, Garrett MD, et al. Trans-activation of the DNAdamage signalling protein kinase Chk2 by T-loop exchange. EMBO J 2006; 25:3179 - 3190
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298:1912 - 1934
- Hanks SK, Hunter T. Protein kinase 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. Faseb J 1995; 9:576 - 596
- Felberg J, Lefebvre DC, Lam M, Wang Y, Ng DH, Birkenhead D, et al. Subdomain X of the kinase domain of Lck binds CD45 and facilitates dephosphorylation. J Biol Chem 2004; 279:3455 - 3462
- Graves PR, Winkfield KM, Haystead TA. Regulation of zipper-interacting protein kinase activity in vitro and in vivo by multisite phosphorylation. J Biol Chem 2005; 280:9363 - 9374
- Haas AL, Bright PM, Jackson VE. Functional diversity among putative E2 isozymes in the mechanism of ubiquitin-histone ligation. J Biol Chem 1988; 263:13268 - 13275
- Haas A, Reback PM, Pratt G, Rechsteiner M. Ubiquitin-mediated degradation of histone H3 does not require the substrate-binding ubiquitin protein ligase, E3 or attachment of polyubiquitin chains. J Biol Chem 1990; 265:21664 - 21669
- Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 2006; 124:1069 - 1081
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, et al. Global mapping of the yeast genetic interaction network. Science 2004; 303:808 - 813
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. The genetic landscape of a cell. Science 2010; 327:425 - 431
- Hartwell LH. Macromolecule synthesis in temperaturesensitive mutants of yeast. J Bacteriol 1967; 93:1662 - 1670
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol 1983; 153:163 - 168
- Pessoa-Brandao L, Sclafani RA. CDC7/DBF4 functions in the translesion synthesis branch of the RAD6 epistasis group in Saccharomyces cerevisiae. Genetics 2004; 167:1597 - 1610
- Foiani M, Liberi G, Piatti S, Plavani P. Cotterill S. Saccharomyces cerevisiae as a model system to study DNA replication. Eukaryotic DNA Replication A Practical Approach 1999; Oxford Oxford University Press 185 - 200
- Shellman YG, Schauer IE, Oshiro G, Dohrmann P, Sclafani RA. Oligomers of the Cdc7/Dbf4 protein kinase exist in the yeast cell. Mol Gen Genet 1998; 259:429 - 436
- Lengronne A, Schwob E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1). Mol Cell 2002; 9:1067 - 1078
- Mann RK, Grunstein M. Histone H3 N-terminal mutations allow hyperactivation of the yeast GAL1 gene in vivo. EMBO J 1992; 11:3297 - 3306