Abstract
ΔNp63α, the dominant negative isoform of the p63 family is an essential survival factor in head and neck squamous cell carcinoma. This isoform has been shown to be down regulated in response to several DNA damaging agents, thereby enabling an effective cellular response to genotoxic agents. Here, we identify a key molecular mechanism underlying the regulation of ΔNp63α expression in response to extrinsic stimuli, such as chemotherapeutic agents. We show that ΔNp63α interacts with NF-κΒ in presence of cisplatin. We find that NF-κΒ promotes ubiquitin-mediated proteasomal degradation of ΔNp63α. Chemotherapy-induced stimulation of NF-κΒ leads to degradation of ΔNp63α and augments trans-activation of p53 family-induced genes involved in the cellular response to DNA damage. Conversely, inhibition of NF-κΒ with siRNA-mediated silencing NF-κΒ expression attenuates chemotherapy induced degradation of ΔNp63α . These data demonstrate that NF-κΒ plays an essential role in regulating ΔNp63α in response to extrinsic stimuli. Our findings suggest that the activation of NF-κΒ may be a mechanism by which levels of ΔNp63α are reduced, thereby rendering the cells susceptible to cell death in the face of cellular stress or DNA damage.
Introduction
The p63 protein is expressed in the basal cells of stratified epithelium such as breast, prostate, skin, cervix and other tissues.Citation1 Recent studies demonstrate the necessity of ΔNp63α in the cellular survival of head and neck squamous cell carcinoma.Citation2 Both p63 and p73 genes encode proteins that possess transactivation, DNA binding and oligomerization domains similar to p53 domains. However, unlike p53, p73 and p63 genes, each contain two independent promoters and make significant use of differential splicing at the gene's 3′ end, hence yielding an array of at least six unique proteins that form two distinct classes: those containing an amino acid terminus (called TAp73 and TAp63) and those that are truncated (called ΔNp73 and ΔNp63) with no amino-terminus region.Citation1,Citation3–Citation6 Both p63 and p73 are homologs of p53 family and are known to be inversely affected by cisplatin. We and others have shown the degradation of ΔNp63α in response to DNA damaging agents like UV, paclitaxel and cisplatin.Citation7,Citation8
The NFκB family of proteins are usually sequestered in the cytoplasm of cells, where it remains inactive through interaction with inhibitory proteins known as IκB.Citation9,Citation10 Stimulation by a variety of extracellular stimuli leads to the phosphorylation of IκB, leading to the activation of NFκB and its translocation into the nucleus. The IκB is there after degraded by the 26S proteasome.Citation11 The two related kinases that phosphorylate the IκB are composed of a 700 kDa kinase complex, whose activities are increased in the presence of stimuli like TNFα and IL-1.Citation12–Citation14 The high molelular complexes are composed of two catalytic subunits IKKβ and IKKα and a regulatory subunit IKKγ (NEMO). Although the degradation of IκB is sufficient to cause nuclear translocation of NFκβ, but other events can affect the NFκB's ability to regulate other proteins. The translocation of NFκB to nucleus has been associated with immune reactions, cell proliferation, apoptotic cell death, tumor proliferation and even ubiquitination.Citation15–Citation19 Also inhibition of NFκB in cancer cells has been associated with tumor proliferation.Citation20 Additionally inhibition of NFκB has been reported to enhance chemotheraupetic sensitivity.Citation21,Citation22 Also, activation of NFκB leads to promotion of apoptosis, depending on the cell type and stimuli.Citation23 Studies reveal that stimulation of NFκB limits the upregulation of p75 in T cells, though the precise mechanism remains to be determined.Citation24 In addition it has been reported that p53 is a direct transcriptional target of NFκB and that inhibition of NFκB activation leads to sequestering of p53 activation signal.Citation25–Citation27 Our recent publication demonstrate that NFκB targeting kinase, IKKβ plays an essential role in regulating ΔNp63α in response to extrinsic stimuli and that the activation of IKK may be a mechanism by which levels of ΔNp63α are reduced, thereby rendering the cells susceptible to cell death in the face of cellular stress or DNA damage.Citation28
In this study we report for the first time that NFκB stimulates the ubiquitin-dependent proteasomal degradation of ΔNp63α. Further our current studies in head and neck cancer cells shows an accumulation of NFκB in the nucleus in response to cisplatin and that ΔNp63α physically interacts with NFκB in presence of Cisplatin. Ectopic expression of NFκB led to a decrease in the levels of ΔNp63α. Further, reduction of NFκB by RNAi resulted in a significant attenuation of the cisplatin-mediated degradation of ΔNp63α. Taken together, this data suggest that NFκB participates in the regulation of the steady-state level of ΔNp63α and may be one of the major determinants of cellular response to cisplatin.
Results
NFκB/p65 interacts with and regulates ΔNp63α in response to cisplatin.
We examined the effect of cisplatin on expression of ΔNp63α in JHU-022 HNSCC cells. Treatment of cells with cisplatin resulted in decreased levels of ΔNp63α within 8 h of exposure (). The loss of ΔNp63α in response to cisplatin was associated with concomitant accumulation of NFκB/p65 in the nuclear fractions of treated cells (). This inverse relationship raised the possibility that activation of NFκB/p65 may play a role in the reduction of ΔNp63α in response to cisplatin treatment. To explore this notion, we investigated whether ΔNp63α physically interacts with either NFκB in JHU-022 cells transfected with expression vectors encoding NFκB/p65. Immunoblot analysis with an anti-p63 antibody demonstrated the presence of endogenous ΔNp63α in anti-NFκB/p65 precipitated immune complexes from transfected cell lysates in presence of cisplatin (). Conversely, immunoblot analysis with an anti-NFκB/p65 specific antibody revealed the presence of NFκB/p65 in anti-flag immunoprecipitates derived from lysates of JHU-022 cells transfected with Flag-ΔNp63α in presence of cisplatin (). We could not detect any interaction between ΔNp63α and NFκB in absence of cisplatin (data not shown). The interaction between endogenous ΔNp63α and NFκB/p65 was established by immunoblot analysis of anti-p63 immunoprecipates with an anti-NFκB/p65 antibody (), and further confirmed by immunoblot analysis of anti-NFκB/p65 immunoprecipates with an anti-p63 antibody in presence of cisplatin (). These results confirmed the interaction between ΔNp63α and NFκB/p65 complex.
NFκB/p65 mediates the reduction of ΔNp63α levels by cisplatin.
JHU-022 cells were transfected with expression vectors encoding the wild-type NFκB/p65. Forced overexpression of NFκB/p65 significantly decreased the steady state levels of ΔNp63α in JHU-022 cells (). To determine whether NFκB/p65 directly affects the stability of ΔNp63α, we examined the effect of co-transfecting a p63- and p53-deficient lung cancer cell line (H1299) with an expression vector encoding ΔNp63α and an expression vector encoding NFκB/p65. Forced expression of NFκB/p65, resulted in a dose-dependent decrease in levels of expression of exogenous ΔNp63α (). These data indicate that NFκB/p65 directly reduces the stability and/or promotes the degradation of ΔNp63α.
To determine whether activation of NFκB/p65 is required for the reduction of endogeous ΔNp63α, we examined the effect of silencing the expression of NFκB/p65 with expression vectors encoding either NFκB/p65 RNAi. Transfection of JHU-022 cells with NFκB/p65 RNAi plasmid, led to significant restoration of ΔNp63α basal expression levels (). To determine whether NFκB/p65 is required for the degradation of ΔNp63α in response to cisplatin, endogenous NFκB/p65 was knocked down with transfection of the respective NFκB/p65 RNAi plasmid. Inhibition of NFκB/p65 expression by RNAi attenuated the degradation of ΔNp63α in response to cisplatin ().
NFκB/p65 promotes ubiquitin-mediated proteasomal degradation of ΔNp63α.
Phosphorylation of substrates, such as IκB proteins, by the IKK complex targets them for ubiquitin-mediated proteasomal degradation. We recently showed that IKKβ promotes proteosomal degradation of ΔNp63α.Citation28 To determine whether NFκB, a downstream target of IKKβ promotes proteasomal degradation of ΔNp63α, JHU-022 cells were transfected with an expression vector encoding NFκB/p65, and then either treated with the proteasome inhibitor, MG132 or left untreated. Whereas expression of NFκB/p65 via transfection resulted in loss of endogenous ΔNp63α, treatment of transfected cells with MG132 significantly attenuated NFκB/p65—mediated degradation of ΔNp63α (). These data suggested that the reduction of ΔNp63α by NFκB/p65 may involve proteasome-mediated degradation. To directly assess whether NFκB/p65 reduces the stability of ΔNp63α protein, we analyzed the effect of expressing NFκB/p65 on the half-life of ΔNp63α in JHU-022 cells. Cells were co-transfected with a plasmid encoding ΔNp63α and an expression vector encoding NFκB/p65, and then exposed to cycloheximide at 24 h following transfection. Immunoblot analysis of ΔNp63α at serial time intervals showed that expression of NFκB/p65 decreased the half-life of ΔNp63α (). To confirm the effect of NFκB/p65 on the half-life of ΔNp63α, JHU-022 cells were transfected with a control vector or NFκB/p65 RNAi vector, and exposed to cycloheximide 24 h following transfection. Immunoblot analysis of ΔNp63α at serial time points showed that the NFκB/p65 silencing by RNAi prolongs the half-life of ΔNp63α ().
Since NFκB/p65 interacts with ΔNp63α and promotes its degradation, we next investigated whether the ubiquitin-proteasome pathway is involved in this process. JHU-022 cells were co-transfected with Flag-ΔNp63α and Ub-HA expression plasmids, with or without increasing concentrations of an expression vector encoding NFκB/p65. At 36 h following transfection, cells were treated with MG132 for 10 h. Cell lysates were immunoprecipitated with anti-HA-matrix and subjected to western blot analysis with an antibody that recognizes ΔNp63α. Expression of NFκB/p65 resulted in a dose-dependent elevation of the ubiquitination of ΔNp63α protein ( and compare lane 2 to lanes 3 and 4). These data demonstrate that NFκB/p65 promotes ubiquitin-mediated degradation of ΔNp63α.
NFκB/p65 counteracts the repressive transcriptional activity of ΔNp63α.
Since NFκB/p65 promoted the ubiquitin-mediated degradation of ΔNp63α, this raised the possibility that NFκB/p65 may enhance p53-dependent transactivation of genes. We investigated the effect of NFκB/p65 on the repressive transcriptional activity of ΔNp63α on two well known targets, p21(WAF1/CIP1) and BAX in JHU-022 cells. Forced expression of ΔNp63α via introduction of the ΔNp63α expression plasmid resulted in a significant reduction of the activity of Bax and p21WAF1 reporter plasmids in JHU-022 cells (). However, co-transfection of an expression vector encoding NFκB/p65 counteracted the repression of either promoter by ΔNp63α (). As previously noted, JHU-022 cells that were transfected with the NFκB/p65 expression plasmid expressed very low levels of endogenous ΔNp63α; forced overexpression of NFκB/p65 in these cells was paralleled by an increase in levels of ΔNp63α target proteins such as Bax and p21WAF1 (). To determine the molecular mechanism by NFκB contributes to the degradation of ΔNp63α , we focused on the transactivation domain of NFκB/p65. Deletion mutant of NFκB/p65Δ450 was used, which lacked the activation domain but retained the nuclear localization domain. Forced expression of NFκB/p65Δ450 did not affect the activity of either the Bax or the p21WAF1 reporter plasmids (). We further examined the effect of NFκB/p65Δ450 on the steady state levels of ΔNp63α in JHU-022 cells, which remained unaffected (). Consistent with their respective effects on the activity of Bax and p21WAF1 reporter plasmids, forced expression of NFκB/p65Δ450 did not affect the expression of endogenous p21 and Bax proteins (), suggesting that the transactivation property of NFκB/p65 is required for degradation of ΔNp63α.
Discussion
p63 activity depends on its steady state protein level and number of evidences suggests that post-translation regulation of the protein plays a major role in response to chemotheraupetic drugs.Citation7,Citation8 Inhibition of ΔNp63α by lentivirus RNAi in HNSCC cells induces the expression of pro-apoptotic genes, Puma and Noxa, suggesting that ΔNp63α is required for cell survival.Citation2 As JHU-022 cells overexpresses ΔNp63α and undetectable amounts of TAp63α,Citation28 our current study is limited to ΔNp63α only. We observed a decrease in ΔNp63α protein levels on cisplatin treatment and a reversion of the same when treated with MG132.Citation7 We demonstrate here a physical interaction between NFκB/p65 and ΔNp63α in the presence of cisplatin. We further report that NFκB/p65 promotes ubiquitin-dependent proteosomal turnover of ΔNp63α. The cisplatin-mediated degradation of ΔNp63α was dependent on NFκB/p65 as evidenced by the RNAi mediated knock down of NFκB/p65 whereby knockdown of NFκB/p65 by RNAi attenuated the cisplatin mediated degradation of ΔNp63α. In this study we found that cisplatin treatment leads to accumulation of NFκB/p65 in the nucleus. Our current findings therefore entail both a novel role of NFκB/p65 in regulating the DNA damage response and a new regulatory pathway of ΔNp63α.
Studies have demonstrated that IKKβ and not IKKα is largely responsible for cytokine induced IκB degradation and nuclear translocation of NFκB. Studies also indicate that activation of NFκB leads to significant instability of p73.Citation16 Various anti-cancer agents like camptothecin, doxorubicin and paclitaxel have been shown to significantly promote nuclear translocation of NFκB.Citation29–Citation31 According to our present study, cisplatin leads to an endogenous activation of NFκB resulting in the significant decrease in the stability of ΔNp63α. We could detect a physical interaction between NFκB/p65 and ΔNp63α only in the presence of cisplatin, which supports our observation of cisplatin mediated translocation of NFκB to nucleus. But even in the absence of cisplatin we observe the steady state level of ΔNp63α affected by forced overexpression of NFκB, which might be through an indirect mechanism. This is in agreement with the observation of Kikuchi et al., where they report NFκB dependent ubiquitin-dependent proteasomal degradation of p73 in the absence of any physical interaction between the two proteins.Citation16 IKKβ has been shown to phosphorylate Serine 536 in the transactivation domain of NFκB/p65, which is necessary for activation of NFκB.Citation32 Deletion of the transactivation domain of NFκB/p65 failed to affect the steady state level of ΔNp63α, raising the possibility that the transactivation domain of NFκB might be required for degradation of ΔNp63α. It has been well established that IKKβ is required for both NFκB liberation and p65 phosphorylation, where as IKKα is solely required for cytokine-induced phosphorylation and activation of NFκB/p65.Citation33 It is likely that IKKβ is critical for NFκB activation and not IKKα. Studies indicate that IKKβ physically binds to ΔNp63α both in the presence of cisplatin and decreases the steady state level of the protein.Citation28
ΔNp63α represses transactivation of both the p21 and Bax promoter-luciferace vector.Citation7,Citation8 We show here that presence of NFκB (which we demonstrate decreases ΔNp63α levels) further enhances the promoter-luciferace activity of both p21 and Bax. It is becoming evident that not all cellular proteins are subjected to ubiquitination-dependent protesomal turnover. It is thus clear that the degradation/stabilization mechanism of ΔNp63α is different from those of p73 and p53, indicating that another as yet unknown pathway may target ΔNp63α. Finally, the ability of NFκB to affect the steady state level of ΔNp63α in response to a chemotherapeutic drug is a novel mechanism that may regulate cellular survival and death to different cellular stress.
Materials and Methods
Plasmids.
ΔNp63α-Flag was by obtained PCR amplification of ΔNp63α cDNA and cloned into the BamHI and NotI sites of pCDNA3.1/hygro (Invitrogen, Carlsbad, CA). NFκB plasmid pCMV4/p65 and pCMV4/p65Δ450 were kindly provided by Dr. Green (Gladstone Institute of Virology and Immunology, UCSF, CA). Plasmid HA-Ub construct was kindly provided by Dr. Bohmann (University of Rochester, NY). Bax-Luciferace expression vector was kindly provided by Dr. John Reed (Burnham Institute for Medical Research, CA) and p21WAF1-Luciferace construct was a kind gift from Prof. Bert Vogelstein (Johns Hopkins University, Baltimore, MD). All clones were sequenced to rule out any mutation.
Cell cultures and transfection.
Human head and neck cancer cell line JHU 022 and Human lung carcinoma cell line (H1299) were maintained in RPMI supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA), 100 U/ml penicillin and 100 µg/ml streptomycin. Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. H1299 and JHU 022 cells were transiently transfected with the indicated expression plasmids using FuGENE HD transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN) in accordance with the manufacturer's specifications. NFκB, RNAi were obtained from Imgenex (San Diego, CA). 48 h post-transfection the cells were treated with or without 75 µM cisplatin before harvesting them for cellular lysates.
Antibodies and immunoblot analysis.
Monoclonal anti-p63 (4A4), monoclonal anti-HA, polyclonal NFκB/p65, antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), monoclonal anti-FLAG (M2) and anti-β-Actin antibodies were obtained from Sigma Chemicals (St. Louis, MO). Western blot was performed as described in reference Citation7. Nuclear and cytoplasmic fractions were prepared as described in reference Citation14.
Immunoprecipitation analysis.
Cells were transfected with various constructs and after 48 h were washed with PBS and lysed using TRITON-X lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM NaCl, 1 mM EDTA; 1% TRITON-X100) containing protease and phosphatase inhibitor cocktails (Sigma Chemical Co.). Lysates were pre-cleaned with protein A-sepharose beads and then incubated for 2 h at 4°C with primary antibody or affinity matrix. The immune complexes were precipitated with protein A-Sepharose beads for 4 h at 4°C, and the nonspecific bound proteins were removed by washing the beads with the NP-40 lysis buffer three times at 4°C. The beads were loaded in Laemmli sample buffer directly and analyzed by immunoblotting with indicated antibody.
Ubiquitination assay.
022 cells were transiently co-transfected with the expression plasmid for HA-tagged ubiquitin, and Flag-ΔNp63α together with or without increasing amounts of NFκB/p65 expression plasmid as indicated. Total amounts of transfected DNA were kept constant by pcDNA3.1 (Invitrogen) per transfection. 48 h after transfection, cells were treated with 25 µM MG-132 and collected in lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM NaCl, 1 mM EDTA; 1% TRITON-X100) containing protease and phosphor inhibitors (Sigma Chemical Co.). Ubiquitinated proteins were recovered by HA resin (Sigma) and analyzed by immunoblotting with the anti-Flag or anti-p63 antibody.
Measurement of p63 half-life.
Cycloheximide (100 µg/ml) (Sigma) was added to 022 cells 24 h after transfection with the indicated combination of plasmids. Protein levels were determined by collecting cells at the indicated time points and immunoblotting was performed as described above. The membrane was re-probed with β-Actin to confirm for equal loading.
Promoter luciferase activity assays.
022 cells were transiently transfected with the Bax and p21WAF1 reporter plasmids along with 500 ng of ΔNp63α expression plasmid (where indicated) in the presence or absence of 1.0 µg of NFκB/p65 or NFκB/p65Δ450 expression plasmid and the empty pGL3-Basic vector were using FuGENE HD according to the manufacturer's protocol (Roche, Indianapolis, IN). DNA concentration in each transfection was kept constant using pCDNA3.1 plasmid. (Invitrogen, Carlsbad, CA). The transfection efficiency was determined by the Renilla luciferase gene-containing pRL-CMV plasmid (Promega, Madison, WI). 48 h after transfection, cells were washed twice with phosphate-buffered saline (PBS) and lysed in a lysis buffer (5x PLBR, Promega, Madison, WI) with gentle shaking at room temperature for 20 min. Cell lysates were centrifuged at 13,000 rpm on a table top centrifuge for 2 min to pellet the cell debris. The supernatants were transferred to a fresh tube, and the dual luciferase activity in cell extracts was determined according to the manufacturer's protocol (Promega). Briefly, each assay mixture contained 2 µl (2 µg) of the cell lysates and 10 µl of a firefly luciferase-measuring buffer (LAR ll R, Promega, Madison, WI). The firefly luciferase activity was measured by a luminometer (the luminometer was programmed to perform a 2 s pre-measurement delay, followed by a 10 s measurement period for each reporter assay). After measuring the firefly luciferase activity (Stop & GloR, Promega, Madison, WI), a Renilla luciferase-measuring buffer was added, and the Renilla luciferase activity was then measured. Each transfection was performed in duplicate, and all experiments were repeated at least three times.
Abbreviations
| Hi | = | histidine |
| JHU | = | Johns Hopkins University |
| NFκB | = | nuclear factor kappaB |
Figures and Tables
Figure 1 NFκB/p65 interacts with and regulates ΔNp63α in response to cisplatin. (A) Loss of ΔNp63α and nuclear accumulation of NFκB/p65 in JHU-022 cells in response to treatment with cisplatin. At the indicated time periods after treatment with cisplatin, JHU-022 cells were fractionated into nuclear and cytoplasmic fractions and analyzed by immunobloting with the indicated antibodies. (B) Complex formation between ΔNp63α with NFκB/p65. (i) JHU-022 cells were transfected with NFκB/p65 or empty Flag-vector and treated with cisplatin or vehicle as indicated. Whole cell lysates were immunoprecipitated with either anti-flag or anti-NFκB/p65 matrix, as indicated, and subjected to western blot analysis using anti-p63 antibody, as indicated. (ii) JHU-022 cells were transfected with Flag-ΔNp63α or empty Flag-vector and treated with cisplatin or vehicle as indicated. Immunoprecipitation was performed using anti-flag matrix and the membrane was blotted with NFκB/p65 antibody, as indicated. (C) Immunoprecipitates of JHU-022 cell lysates using either anti-p63 (i) or anti-NFκB/p65 (ii) antibody were subjected to immunoblot analysis using either anti-NFκB/p65 or anti-p63 antibody, as indicated.
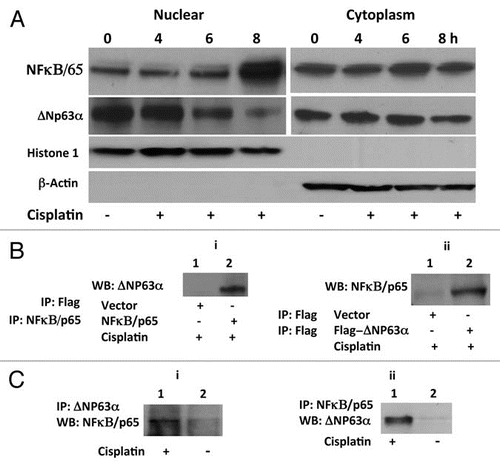
Figure 2 NFκB/p65 mediates the reduction of ΔNp63α levels by cisplatin. (A and B) JHU-022 and p53/p63 deficient H1299 cells were transfected with increasing concentrations (0, 0.5, 1 and 1.5 µg) of NFκB/p65 expression plasmids as indicated; western blot was performed using the indicated antibodies. p53/p63 deficient H1299 cells were also transfected with 1 µg of ΔNp63α expression plasmid. (C) JHU-022 cells were transfected with increasing concentrations of either NFκB/p65 RNAi plasmid and subjected to western blot analysis using anti-p63 antibodies to assess the endogenous levels of p63. (D) JHU-022 cells were transfected with or without NFκB/p65 plasmid 24 h after transfection, the cells were treated with or without 75 µM cisplatin for 8 h, and cell lysates were subjected to western blot using anti-p63 antibodies.
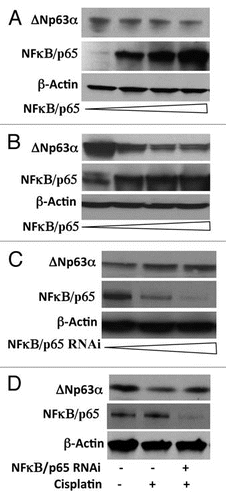
Figure 3 NFκB/p65 promotes ubiquitin-mediated proteasomal degradation of ΔNp63α. (A) JHU-022 cells were transfected with or without an expression plasmid encoding NFκB/p65; 24 h after transfection, cells were treated with 10 µM MG132 for the indicated time periods. Whole cell lysates were subjected to western Blot analysis using anti-NFκB/p65 or anti-p63 antibodies. (B) JHU-022 cells were transfected with constant amount of ΔNp63α expression plasmid with or without expression plasmids encoding NFκB/p65, or empty vector, as indicated. 24 h after transfection, the cells were treated with 100 µg/ml cycloheximide. At the indicated time points, whole cell lysates were analyzed for ΔNp63α by immunoblotting. Actin was used for loading control. (C) JHU-022 cells were transfected with constant amount of NFκB/p65 RNAi expression plasmid. 24 h after transfection, the cells were treated with 100 µg/ml cycloheximide. At the indicated time points, whole cell lysates were analyzed for endogenous ΔNp63α by immunoblotting. Actin was used for loading control. (D) JHU-022 cells were co-transfected with ΔNp63α and Ub-HA expression plasmids, with or without increasing concentrations of an expression vector encoding NFκB/p65. At 36 h following transfection, cells were treated with MG132 for 10 h. Cell lysates were immunoprecipitated with anti-HA-matrix and subjected to western blot analysis with an antibody that recognizes ΔNp63α to assess the ubiquitination levels of ΔNp63α.
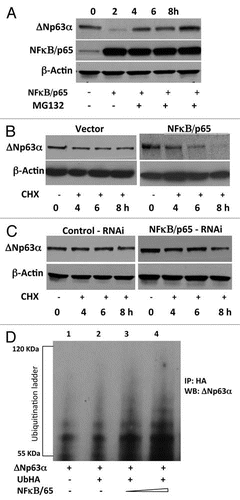
Figure 4 NFκB/p65 counteracts the repressive transcriptional activity of ΔNp63α. (A) JHU-022 cells were transfected with either p21 or Bax luciferase promoter construct along with Renilla luciferase plasmid, with or without NFκB/p65 or NFκB/p65Δ450 and/or ΔNp63α, as indicated. The amount of DNA per transfection was kept constant by using empty pCDNA3.1 vector. At 24 h post-transfection, the luciferase activity was determined. The transfection efficiency was standardized against Renilla luciferase. Results shown are representative of three independent experiments. *indicated p ≤ 0.001. (B) JHU-022 cells were transfected with increasing concentrations of NFκB/p65 expression plasmid and western blot analysis was performed with the indicated antibodies to assess the endogenous levels of the indicated proteins. (C) (i) JHU-022 cells were transfected with empty vector or expression vector encoding NFκB/p65Δ450, and western blot analysis was performed with the indicated antibodies to assess the endogenous levels of the indicated proteins. (ii) A diagramatic representation of the NFκB/p65 domains.
Acknowledgements
This research was supported by FAMRI-funded Young Clinical Scientist Award 072017_YCSA, titled “The effect of Adenylate Kinase 3 (AK3) on tobacco smoke-induced cisplatin resistance in bladder cells” and by National Institutes of Health Grant 5-R01 DE 13561, titled “The Role of P40 Squamous Cell Carcinoma.”
References
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing and dominant-negative activities. Mol Cell 1998; 2:305 - 316
- Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006; 9:45 - 56
- Benard J, Douc-Rasy S, Ahomadegbe JC. TP53 family members and human cancers. Hum Mutat 2003; 21:182 - 191
- Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S. Possible oncogenic potential of DeltaNp73: A newly identified isoform of human p73. Cancer Res 2002; 62:636 - 641
- Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997; 389:191 - 194
- Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M, et al. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is upregulated in human tumors. J Exp Med 2002; 196:765 - 780
- Chatterjee A, Upadhyay S, Chang X, Nagpal JK, Trink B, Sidransky D. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 2008; 7:1231 - 1237
- Westfall MD, Joyner AS, Barbieri CE, Livingstone M, Pietenpol JA. Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of DeltaNp63alpha. Cell Cycle 2005; 4:710 - 716
- Baeuerle PA, Baltimore D. NFkappaB: Ten years after. Cell 1996; 87:13 - 20
- Beg AA, Finco TS, Nantermet PV, Baldwin AS Jr. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IkappaBalpha: a mechanism for NFkappaB activation. Mol Cell Biol 1993; 13:3301 - 3310
- Rothwarf DM, Karin M. The NFkappaB activation pathway: A paradigm in information transfer from membrane to nucleus. Sci STKE 1999; 1999:1
- Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, et al. Signal-induced site-specific phosphorylation targets IkappaB alpha to the ubiquitin-proteasome pathway. Genes Dev 1995; 9:1586 - 1597
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NFkappaB. Nature 1997; 388:548 - 554
- Rothwarf DM, Zandi E, Natoli G, Karin M. IKKgamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998; 395:297 - 300
- Beg AA, Baltimore D. An essential role for NFkappaB in preventing TNFalpha-induced cell death. Science 1996; 274:782 - 784
- Kikuchi H, Ozaki T, Furuya K, Hanamoto T, Nakanishi M, Yamamoto H, et al. NFkappaB regulates the stability and activity of p73 by inducing its proteolytic degradation through a ubiquitin-dependent proteasome pathway. Oncogene 2006; 25:7608 - 7617
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNFalpha-induced apoptosis by NFkappaB. Science 1996; 274:787 - 789
- Wang CY, Mayo MW, Baldwin AS Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NFkappaB. Science 1996; 274:784 - 787
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NFkappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998; 281:1680 - 1683
- Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NFkappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 2004; 6:297 - 305
- Cusack JC Jr, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, et al. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res 2001; 61:3535 - 3540
- Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: Enhanced anti-tumor therapy through increased apoptosis by inhibition of NFkappaB. Nat Med 1999; 5:412 - 417
- Huang Y, Fan W. IkappaB kinase activation is involved in regulation of paclitaxel-induced apoptosis in human tumor cell lines. Mol Pharmacol 2002; 61:105 - 113
- Wan YY, DeGregori J. The survival of antigen-stimulated T cells requires NFkappaB-mediated inhibition of p73 expression. Immunity 2003; 18:331 - 342
- Hellin AC, Calmant P, Gielen J, Bours V, Merville MP. Nuclear factorkappaB-dependent regulation of p53 gene expression induced by daunomycin genotoxic drug. Oncogene 1998; 16:1187 - 1195
- Sun X, Shimizu H, Yamamoto K. Identification of a novel p53 promoter element involved in genotoxic stress-inducible p53 gene expression. Mol Cell Biol 1995; 15:4489 - 4496
- Wu H, Lozano G. NFkappaB activation of p53. A potential mechanism for suppressing cell growth in response to stress. J Biol Chem 1994; 269:20067 - 20074
- Chatterjee A, Chang X, Sen T, Ravi R, Bedi A, Sidransky D. Regulation of p53 family member isoform DeltaNp63alpha by the nuclear factorkappaB targeting kinase IkappaB kinase beta. Cancer Res 70:1419 - 1429
- Bian X, McAllister-Lucas LM, Shao F, Schumacher KR, Feng Z, Porter AG, et al. NFkappaB activation mediates doxorubicin-induced cell death in N-type neuroblastoma cells. J Biol Chem 2001; 276:48921 - 48929
- Huang TT, Wuerzberger-Davis SM, Seufzer BJ, Shumway SD, Kurama T, Boothman DA, et al. NFkappaB activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J Biol Chem 2000; 275:9501 - 9509
- Huang Y, Johnson KR, Norris JS, Fan W. Nuclear factorkappaB/IkappaB signaling pathway may contribute to the mediation of paclitaxel-induced apoptosis in solid tumor cells. Cancer Res 2000; 60:4426 - 4432
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NFkappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 1999; 274:30353 - 30356
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the IkappaB kinase alpha and beta subunits in liberating nuclear factor kappaB (NFkappaB) from IkappaB and in phosphorylating the p65 subunit of NFkappaB. J Biol Chem 2002; 277:3863 - 3869
- Chatterjee A, Mambo E, Osada M, Upadhyay S, Sidransky D. The effect of p53-RNAi and p53 knockout on human 8-oxoguanine DNA glycosylase (hOgg1) activity. Faseb J 2006; 20:112 - 114