Abstract
Mutations in the MCPH1 gene cause primary microcephaly associated with a unique cellular phenotype of misregulated chromosome condensation. The encoded protein contains three BRCT domains, and accumulating data show that MCPH1 is involved in the DNA damage response. However, most of this evidence has been generated by experiments using RNA interference (RNAi) and cells from non-human model organisms. Here, we demonstrate that patient-derived cell lines display a proficient G2/M checkpoint following ionizing irradiation (IR) despite homozygous truncating mutations in MCPH1. Moreover, chromosomal breakage rates and the relocation to DNA repair foci of several proteins functioning putatively in an MCPH1-dependent manner are normal in these cells. However, the MCPH1-deficient cells exhibit a slight delay in re-entering mitosis and delayed resolution of γH2AX foci following IR. Analysis of chromosome condensation behavior following IR suggests that these latter observations may be related to hypercondensation of the chromatin in cells with MCPH1 mutations. Our results indicate that the DNA damage response in human cells with truncating MCPH1 mutations differs significantly from the damage responses in cells of certain model organisms and in cells depleted of MCPH1 by RNAi. These subtle effects of human MCPH1 deficiency on the cellular DNA damage response may explain the absence of cancer predisposition in patients with biallelic MCPH1 mutations.
Introduction
Autosomal recessive primary microcephaly (MCPH, OMIM 251200) is a genetically heterogeneous neurodevelopmental disorder characterized by markedly reduced brain size and mental retardation. Mutations in one of the underlying genes, MCPH1 (alternatively termed BRIT1), cause primary microcephaly associated with a unique cellular phenotype of defective chromosome condensationCitation1–Citation3 (OMIM 606858). Proliferating cultures of cells with MCPH1 mutations contain substantially more cells with a prophase-like appearance (prophase-like cells, PLCs); the chromatin of these cells is highly condensed within a retained nuclear membrane. We have shown that this increase is due to premature chromatin condensation in the G2 phase of the cell cycle and to delayed decondensation in the early G1 phase.Citation1,Citation3 We have also demonstrated that these condensation defects are caused by misregulation of the condensin II protein complex.Citation4
The MCPH1 protein contains three BRCT domains. In addition to its function in the regulation of chromosome condensation, MCPH1 seems to be involved in the maintenance of centrosomal integrity and in the DNA damage response (reviewed in ref. Citation5). However, evidence that MCPH1 participates in the DNA damage response comes mainly from experiments using RNA interference (RNAi) in established human cancer cell lines and from Mcph1 knock-out mice cells. A severe impairment of the DNA damage-induced G2/M checkpoint in the human osteosarcoma cell line U2OS was reported after RNAi-mediated depletion of MCPH1.Citation6,Citation7 In addition, the protein levels of the checkpoint mediator BRCA1 and the checkpoint kinase CHK1 decreased following treatment with MCPH1 siRNA. Rai et al.Citation8 reported that MCPH1 co-localizes with DNA damage response proteins such as MDC1, 53BP1, NBS1 and phosphorylated ATM and that RNAi against MCPH1 impairs the targeting of these proteins to IR-induced foci (IRIF). It was also demonstrated that the ability of MCPH1 to localize to the sites of DNA double-strand breaks (DSBs) depends on its C-terminal tandem BRCT domains.Citation9,Citation10 In addition, it was shown that functional crosstalk between MCPH1 and condensin II is required not only for the regulation of chromosome condensation but also for homologous repair of DNA damage.Citation11 Therefore, it was postulated that MCPH1 might be a tumor suppressor gene. Consistent with this hypothesis the copy number and expression of MCPH1 were found to be reduced in several types of tumors, and MCPH1 expression was found to be inversely correlated with genomic instability and metastasis.Citation8,Citation12
Moreover, it was recently observed that Mcph1 is essential for DNA repair and the maintenance of genomic stability in mice.Citation13 Mcph1 knock-out mice and cells derived from these animals were found to be hypersensitive to IR, and all Mcph1-/- mice died within nine days after irradiation at a dose at which 80% of wild-type animals were still alive four weeks later. Moreover, murine Mcph1-/- embryonic fibroblasts and T lymphocytes exhibited increased chromosomal breakage following irradiation.
Most MCPH1 patients reported to date bear homozygous early truncating mutations that should result in a loss of function of the gene products. Thus, one would expect that the consequences of these mutations would resemble those described in model organisms and, on the cellular level, those induced by RNAi-mediated depletion of MCPH1. Mutations in the DNA damage response genes NBS1 and ATM result in the disorders Nijmegen breakage syndrome (NBS) and ataxia teleangiectasia (AT), respectively. Cells from these patients display defects in the response to IR similar to the defects described for cells treated with siRNA against MCPH1 and for Mcph1-/- mice. Consequently, NBS and AT patients have a strong predisposition for malignancies. Moreover, patients with AT and NBS are unusually sensitive to X-rays, and treatment of malignancies with conventional dosages of radiation can be fatal to these patients. Until now, there have been no reports about the response of the cells of MCPH1 patients to IR. Aditionally, there have been no reports about malignancies in MCPH1 patients. With regard to the clinical management of MCPH1 patients, it is therefore important to analyze the response of their cells to IR and to compare this response to those observed in Mcph1-/- mice and siRNA-treated cells.
Results
Chromosomal breakage.
To analyze the ability of the cells of MCPH1 patients to repair DNA DSBs, we exposed lymphoblastoid cell lines (LCLs) derived from five controls and from five MCPH1 patients bearing three different homozygous mutations (c.427dupA/p.T143NfsX5, c.74C>G/p.S25X, c.223T>C/p. W75R) to IR at 0.5 or 1 Gy and prepared chromosomes 6 h and 24 h later. The slides were coded, and metaphase cells were screened microscopically for chromosomal breakage. As shown in , there was no increase in the chromosomal breakage rate or in the proportion of aberrant metaphase cells in the patient cells compared to the controls.
Proficient G2/M checkpoint control in MCPH1 patient cells.
We have shown previously that irradiation of MCPH1 patient cells does not result in disproportional G2 phase accumulation,Citation1 but several groups have reported that knockdown of MCPH1 by RNAi results in the loss of transient G2/M arrest following exposure to IR.Citation6,Citation7 Thus far, it has not been determined whether MCPH1 patient cells display a functional early G2/M checkpoint arrest, which prevents cells with damaged DNA from entering into mitosis. To investigate their G2/M checkpoint control, LCLs from patients with different homozygous MCPH1 mutations (one missense mutation, c.80C>G/p.T27R; and two different truncating mutations, c.427dupA/p.T143NfsX5 and c.74C>G/p. S25X) were exposed to IR at 1 Gy together with two normal control LCLs and a cell line derived from an AT patient. The cells were assayed 2 h later by flow cytometry to determine their mitotic indices. Under these conditions, all MCPH1 patient cells arrested in G2/M phase to an extent comparable to that of normal control cells (). The mitotic index was reduced from 1.4% to 0.1% in the MCPH1 patient cells and from 1.5% to 0.1% in the normal control cells. In contrast, in the AT cell line, the mitotic index was reduced from 1.3% to only 0.68% following IR. This result demonstrates that the MCPH1 patient cells were G2/M checkpoint proficient, whereas the AT cells proceeded into mitosis despite the DNA damage induced by IR.
MCPH1 patient cells demonstrate delayed release from G2/M checkpoint arrest.
As an indicator of DNA damage repair efficiency, we investigated the time required for cells to release from the checkpoint after IR and enter mitosis. In this experiment, exponentially growing LCLs were irradiated with 1 Gy, and the mitotic index was determined at 1 h intervals for 8 h. Again, in contrast to the AT cells, the MCPH1 patient cells exhibited an efficient G2/M checkpoint arrest similar to that of the normal controls (). However, we observed a delay in checkpoint release. The MCPH1 patient cells recovered from the checkpoint arrest with a slight but clear and reproducible delay compared to the control cells. While normal control cells released from checkpoint arrest 4 h after IR, MCPH1 cells recovered 5 h after IR, as shown by the mitotic indices ().
This delayed checkpoint release suggested that the completion of DSB repair was likewise delayed. We therefore monitored the persistence of phosphorylated H2AX (γH2AX) in IRIF which correlates with the presence of unrepaired DNA DSBs.Citation14 We determined the number of cells displaying γH2AX IRIF sequentially post-IR among MCPH1 patient and control cells. The MCPH1 patient cells formed γH2AX IRIF as efficiently as the normal control cells did (). However, the proportion of γH2AX IRIF-positive cells decreased more slowly in the MCPH1 cells compared to the control cells, suggesting that the persistence of DSBs may cause the delayed checkpoint release in MCPH1 patient cells.
Effect of MCPH1 mutations on ionizing irradiation-induced foci formation.
It has also been reported that certain DNA damage response proteins are targeted to DNA repair foci at the sites of DSBs in an MCPH1-dependent manner.Citation8 Therefore, we explored whether the MCPH1 mutations interfere with focus formation by important downstream targets following DSB induction. HeLa cells and fibroblasts established from a patient with a homozygous truncating MCPH1 mutation (c.427dupA/p.T143NfsX5) were irradiated with 10 Gy, fixed 1 h later, and labeled with antibodies against 53BP1, RAD51 and NBS1. Surprisingly, all of the tested proteins formed IRIF at normal rates (). Importantly, there were no significant differences in the proportion of foci-positive cells between the normal control (HeLa), the MCPH1-mutated fibroblasts, and the isogenic controls (i.e., MCPH1 patient fibroblasts expressing EGFP or complemented by ectopic expression of wild-type MCPH1). Functional complementation of MCPH1 was ascertained by reversion of the condensation defect (data not shown).
Chromatin condensation following ionizing irradiation.
The typical cellular phenotype induced by the loss of MCPH1 function is characterized by premature condensation in the early G2 phase of the cell cycle and delayed decondensation in the early G1 phase,Citation1,Citation3 resulting in a high number of cells with a prophase-like appearance (PLCs). Normal prophase cells decondense their chromatin following DNA damage to allow repair unless they have passed a so-called “point of no return.”Citation15 Recently, it was also shown that MCPH1 regulates chromatin remodeling via the SWI-SNF complex in response to DNA damage to facilitate DNA repair.Citation16 Therefore, we asked whether IR would affect the chromosome condensation behavior of MCPH1 patient cells. Cultures of lymphoblastoid patient cells were exposed to different doses of IR. Cytological preparations were made at 1 h intervals for 8 h following IR to analyze chromatin morphology. While the metaphase indices dropped to approximately zero by 1 h following IR at all doses applied—confirming effective checkpoint control—the proportion of PLCs remained constant (). This result correlates with our finding that IRIF form in cells with hypercondensed chromatin (). Thus, it appears that MCPH1 patient cells do not decondense their chromatin completely following DNA damage. Nevertheless, the damage is recognized, and detector and repair proteins have access to the sites of damage.
Shortly after the cells reentered mitosis, the number of PLCs started to increase above the average level. We hypothesized that this may mirror the cell cycle distribution of the patient cells at this time point post-IR. The increase in the number of cells with a prophase-like appearance may be a consequence of the condensation defect combined with the normal response of the patient cells to DNA damage: the cells accumulate in the G2 phase due to an effective G2/M checkpoint and consequently condense their chromatin. To investigate this hypothesis further, we repeated the experiment described above, but we performed flow cytometric analysis of cell cycle distribution in parallel with the microscopic determination of the proportion of PLCs. Cells were irradiated, fixed, and processed at 2 h intervals for 12 h (). Importantly, the changes in the cell cycle distribution of the patient cells resembled those of the control cells. While the fraction of G1 cells decreased following IR, reaching a minimum at 4 h, the proportion of cells in the S phase reached a maximum at the same time. When cells started to leave the S phase, they accumulated in G2, again indicating the retention of a functional G2/M checkpoint. The parallel microscopy analysis of condensation behavior revealed that the proportion of PLCs reached its maximum at the same time ().
Discussion
It is widely agreed that the DNA damage response network (including proper checkpoint control) is a critical barrier against genomic instability and that shortcomings in this system confer susceptibility to cancer. Several reports attribute a crucial function in DNA damage response and checkpoint control to MCPH1, which would have severe consequences for the health of patients with MCPH1 mutations. Surprisingly, our results show that cells of MCPH1 patients bearing homozygous N-terminal truncating mutations are G2/M checkpoint proficient in response to IR and effectively prevent the progression of damaged cells into mitosis. Moreover, chromosomal breakage rates were not higher in the patient cells compared to control cells following IR, and proteins putatively downstream of MCPH1 localized efficiently to IRIF. Thus, MCPH1 patient cells differ significantly in their response to DNA damage from cells treated with siRNA against MCPH1 or cells from Mcph1-/- mice. These results are in accordance with the clinical phenotype of MCPH1 patients, which is more reminiscent of ATR-Seckel syndrome than disorders with defects in DNA double-strand repair such as NBS or AT.Citation17 Moreover, it has been shown previously that MCPH1 patient cells show defects in ATR-mediated checkpoint responses.Citation18
Nevertheless, the discrepancies concerning the response to IR remain to be explained. Currently we can only speculate as to why our investigations demonstrate that the response to IR in MCPH1 patient cells is largely normal. Incomplete loss of function due to hypomorphic mutations may be an explanation, and it has already been reported that residual protein can be detected in c.74C>G/p.S25X cells.Citation18 However, our own western blot analyses using MCPH1-specific antibodies showed that MCPH1 is virtually unexpressed in these cells (Sup. Fig. 1). Yet, it should be kept in mind that RNAi methods also usually fail to achieve complete depletion of target proteins. The differences may be explained by the observed downregulation of BRCA1 following RNAi against MCPH1. The early G2/M checkpoint is dependent on BRCA1,Citation19,Citation20 and our own unpublished data confirm that the expression levels of BRCA1 are lower neither in patient lymphocytes nor in LCLs (reviewed in ref. Citation18). Thus, it may be possible that the effects observed following RNAi reflect the immediate response to MCPH1 deprivation, while in mutated cells, redundant pathways are upregulated. Even if this interpretation is correct, it would not fully explain the differences with respect to some of the reported transgenic animals.
It should also be noted that the published data from model organisms contain inconsistencies. While severe defects in the DNA damage response were reported for an Mcph1-/- mouse model,Citation13 we were unable to detect these DNA damage response defects in cells derived from mice bearing Mcph1 gene trap mutationsCitation21 resulting in deletion of the C-terminal BRCT domain of the mouse Mcph1. We observed a reduced life span among these Mcph1-deficient mice, but the reasons for the premature death of these animals remain elusive. Furthermore, cells from Drosophila mcph1-mutant larvae exhibit IR-induced G2 arrest,Citation22 and while we were preparing our manuscript, observations similar to ours were published after disruption of Mcph1 in the hyper-recombinogenic DT40 chicken cell line.Citation23 Again, this may be due to the hypomorphicity of the mutations, or it may be that different types of mutations cause divergent effects in various species. To reliably determine the clinical prognosis for MCPH1 patients, it will be important to find definite explanations for these observed differences, and it would be interesting to investigate whether BRCA1 is downregulated in the murine Mcph1-/- cells with a defective DNA damage response.
Our results show that the impact of MCPH1 mutations on the response to DNA damage in patients with primary microcephaly may not be as severe as suggested by experiments with RNAi and Mcph1-/- mice. This discrepancy may explain the absence of cancer predisposition in these patients. However, the delay we detected in checkpoint release demonstrates that the response to IR may be somewhat defective. It is tempting to speculate that repair may be impaired by an unfavorable environment created by the highly condensed chromatin. In accordance with our observations, it has been shown that MCPH1 regulates the ATP-dependent SWI-SNF chromatin remodeling complex during DNA repair.Citation16 This result may also explain the prolonged preservation of γH2AX IRIF and the delayed checkpoint recovery in MCPH1 patient cells. It will be interesting to further investigate chromatin dynamics and DNA repair within this context in real time and on a submicroscopic level.
Furthermore, our results could provide an alternative explanation for the pathogenesis of microcephaly in MCPH1 patients. The delayed release from checkpoint arrest in MCPH1-deficient cells may subtly affect cell division rates. This impairment may not be noticeable in many somatic tissues, but due to the exponential expansion of the progenitor cell pool during neurogenesis, even a subtle perturbation of the cell division rate could cause microcephaly without inducing detectable abnormalities in other tissues.Citation24
Materials and Methods
Cell culture.
Lymphoblastoid cell lines were grown in RPMI-1640 medium (Invitrogen) containing 15% fetal bovine serum (FBS) and maintained at 37°C in 5% CO2. Adherent cell lines were grown in a monolayer in minimum essential medium supplemented with 10% FBS.
Chromosomal breakage analysis.
Logarithmically proliferating lymphoblastoid cell cultures were irradiated using an X-ray source (Muller MG 150, Ua = 100 kV, I = 10 mA, filter 0.3 mm Ni, dose rate: 2.1 Gy/min). Chromosomes were prepared at 6 h and 24 h after the time of irradiation using standard diagnostic laboratory procedures. Slides were stained by immersion in fresh 10% Giemsa stain for 10 min. The slides were coded, and at least 200 metaphase cells were scored for both the patients and the controls at each dose and time point.
Flow cytometry.
Cells were washed in PBS and fixed in 2% paraformaldehyde. The cells were permeabilized with 90% methanol for 30 min on ice. Phospho-histone H3 was detected by a mouse monoclonal anti p-H3 primary antibody (Cell Signaling Technology) at a dilution of 1:25 and a goat anti-mouse IgG (H+L) Alexa Fluor 647-conjugated secondary antibody (Molecular Probes). DAPI was used as a counterstain for DNA content and cell cycle distribution. Fluorescence detection was performed using an analytical flow cytometer (Becton Dickinson, LSRII) equipped with FACSDiva software for data acquisition. Quantitative cell cycle analysis was done with WinMDI software.
Immunofluorescence.
For immunofluorescence, cells grown on coverslips were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 15 min at room temperature and then permeabilized with ice-cold methanol for 30 min on ice. Cells were incubated with PBS containing 20% FBS as a blocking agent for 30 min and then with the indicated antibody (53BP1 (Novus), NBS1 (Abcam) or RAD51 (Abcam)) for approximately 1 h at room temperature. After being washed three times with PBS, the cells were incubated with the respective secondary antibody conjugated with Alexa Fluor 594 (Molecular Probes) for 30 min. DNA was stained with 1 µg/ml DAPI for 10 min at 4°C. Following a PBS rinse, the coverslips were mounted with ProLong antifade reagent (Invitrogen). Fluorescence images were captured and processed using an Axiovert 200M inverted microscope equipped with a Plan Apo 63x/1.4 oil immersion objective and AxioVision software (Carl Zeiss).
Abbreviations
| MCPH | = | autosomal recessive primary microcephaly |
| BRCT | = | BRCA1 carboxy-terminal (domain) |
| LCLs | = | lymphoblastoid cell lines |
| IR | = | ionizing radiation |
| PLCs | = | prophase-like cells |
| IRIF | = | ionizing radiation-induced foci |
| DSBs | = | double-stranded breaks |
Figures and Tables
Figure 1 DNA repair and G2/M checkpoint response of MCPH1 patient cells. (A) To analyze chromosomal breakage rates, lymphoblastoid cell lines (LCLs) derived from MCPH1 patients were exposed to different doses of IR. Metaphase spreads were prepared according to standard protocols at 0 h, 6 h and 24 h following irradiation. The number of chromosomal breaks per cell is shown to the left, and the percentage of aberrant metaphase cells is presented to the right. (B) G2/M checkpoint analysis. MCPH1 patients with the indicated homozygous mutations, LCLs derived from normal controls, and one ATM-mutated cell line (A-T) were irradiated with 1 Gy, and their mitotic indices were determined using flow cytometry by staining of mitotic cells with an antibody to phospho-histone H3. (C) G2/M checkpoint release. Cells were processed and analyzed as described above. Mitotic indices in patient and control LCLs were determined sequentially at the indicated time points after exposure to 1 Gy. (D) Immunofluorescence analysis of γH2AX IRIF in the cells analyzed in (C). The data represent the proportion of cells with >10 γH2AX foci. Error bars indicate the SD.
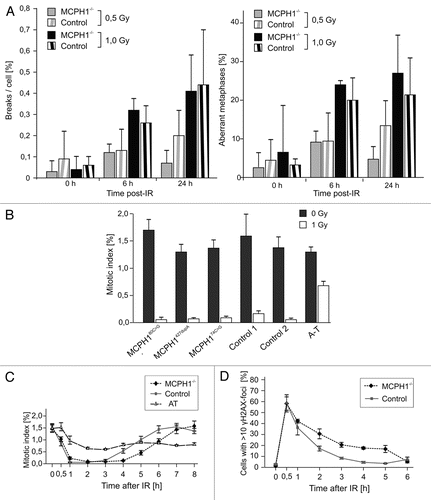
Figure 2 IRIF formation in MCPH1 patient cells. (A) Immunofluorescence micrographs showing 53BP1, NBS1 and RAD51 IRIF (red) in HeLa cells and transformed fibroblasts derived from a patient with a homozygous truncating MCPH1 mutation (c.427dupA/p.T143NfsX5) following irradiation with 10 Gy. The two righthand columns show fibroblasts from the same MCPH1 patient stably expressing EGFP or EGFP fused to wild-type MCPH1. Nuclei were counterstained with DAPI (blue). (B) Quantification of cells with >10 IRIF. Error bars indicate the SD of three different measurements, each numbering approximately 200 nuclei. Scale bar = 5 µm.
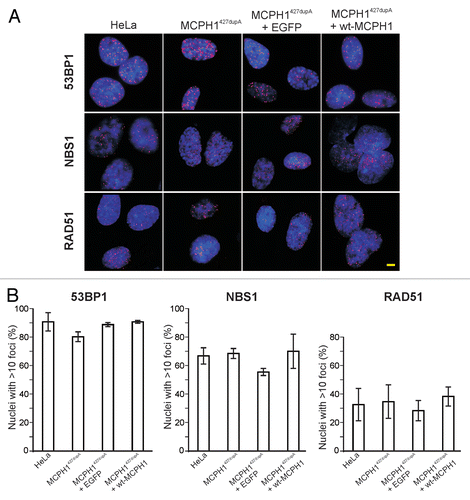
Figure 3 Chromosome condensation behavior and cell cycle distribution in response to IR. (A) LCLs derived from two MCPH1 patients with homozygous truncating mutations were irradiated with 0.5 Gy, 1 Gy or 2 Gy, and cytogenetic slides were prepared at varying times following IR as indicated. The fractions of mitotic and prophase-like cells were determined by counting 1,000 cells per time point. (B) Immunofluorescence micrographs showing 53BP1 IRIF (red) in fibroblasts from an MCPH1 patient with hypercondensed, prophase-like chromatin morphology. Nuclei were counterstained with DAPI (blue). Scale bar = 5 µm. (C) Cell cycle distribution of MCPH1 patient and control LCLs at varying times after 1 Gy IR as determined by flow cytometry. Error bars indicate SD. (D) Determination of the proportion of cells with a prophase-like appearance in the same experiment.
Additional material
Download Zip (257.5 KB)Acknowledgements
We thank Sylke Niehage and Richard Friedl for excellent technical assistance and Andrew Jackson for the 74C>G MCPH1-mutant lymphoblastoid cells. This research was supported by funding from the Deutsche Forschungsgemeinschaft (NE 531/5-3) to H.N.
References
- Neitzel H, Neumann LM, Schindler D, Wirges A, Tonnies H, Trimborn M, et al. Premature chromosome condensation in humans associated with microcephaly and mental retardation: a novel autosomal recessive condition. Am J Hum Genet 2002; 70:1015 - 1022
- Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet 2002; 71:136 - 142
- Trimborn M, Bell SM, Felix C, Rashid Y, Jafri H, Griffiths PD, et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am J Hum Genet 2004; 75:261 - 266
- Trimborn M, Schindler D, Neitzel H, Hirano T. Misregulated chromosome condensation in MCPH1 primary microcephaly is mediated by condensin II. Cell Cycle 2006; 5:322 - 326
- Thornton GK, Woods CG. Primary microcephaly: Do all roads lead to Rome?. Trends Genet 2009; 25:501 - 510
- Xu X, Lee J, Stern DF. Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J Biol Chem 2004; 279:34091 - 34094
- Lin SY, Rai R, Li K, Xu ZX, Elledge SJ. BRIT1/MCPH1 is a DNA damage responsive protein that regulates the Brca1-Chk1 pathway, implicating checkpoint dysfunction in microcephaly. Proc Natl Acad Sci USA 2005; 102:15105 - 15109
- Rai R, Dai H, Multani AS, Li K, Chin K, Gray J, et al. BRIT1 regulates early DNA damage response, chromosomal integrity and cancer. Cancer Cell 2006; 10:145 - 157
- Wood JL, Singh N, Mer G, Chen J. MCPH1 functions in an H2AX-dependent but MDC1-independent pathway in response to DNA damage. J Biol Chem 2007; 282:35416 - 35423
- Jeffers LJ, Coull BJ, Stack SJ, Morrison CG. Distinct BRCT domains in Mcph1/Brit1 mediate ionizing radiation-induced focus formation and centrosomal localization. Oncogene 2008; 27:139 - 144
- Wood JL, Li K, Liang Y, Chen J. Microcephalin/MCPH1 associates with the condensin ii complex to function in homologous recombination repair. J Biol Chem 2008;
- Rai R, Phadnis A, Haralkar S, Badwe RA, Dai H, Li K, et al. Differential regulation of centrosome integrity by DNA damage response proteins. Cell Cycle 2008; 7:2225 - 2233
- Liang Y, Gao H, Lin SY, Peng G, Huang X, Zhang P, et al. BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice. PLoS Genet 2010; 6:1000826
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858 - 5868
- Rieder CL, Cole RW. Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. J Cell Biol 1998; 142:1013 - 1022
- Peng G, Yim EK, Dai H, Jackson AP, Burgt I, Pan MR, et al. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat Cell Biol 2009; 11:865 - 872
- Kerzendorfer C, O'Driscoll M. Human DNA damage response and repair deficiency syndromes: linking genomic instability and cell cycle checkpoint proficiency. DNA Repair 2009; 8:1139 - 1152
- Alderton GK, Galbiati L, Griffith E, Surinya KH, Neitzel H, Jackson AP, et al. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat Cell Biol 2006; 8:725 - 733
- Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol 2001; 21:3445 - 3450
- Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol 2002; 22:1049 - 1059
- Trimborn M, Ghani M, Walther DJ, Dopatka M, Dutrannoy V, Busche A, et al. Establishment of a mouse model with misregulated chromosome condensation due to defective Mcph1 function. PLoS One 2010; 5:9242
- Rickmyre JL, Dasgupta S, Ooi DL, Keel J, Lee E, Kirschner MW, et al. The Drosophila homolog of MCPH1, a human microcephaly gene, is required for genomic stability in the early embryo. J Cell Sci 2007; 120:3565 - 3577
- Brown JA, Bourke E, Liptrot C, Dockery P, Morrison CG. MCPH1/BRIT1 limits ionizing radiationinduced centrosome amplification. Oncogene 2010; 29:5537 - 5544
- Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci 1995; 18:383 - 388