Abstract
In the mammalian CNS, excessive release of glutamate and overactivation of glutamate receptors are responsible for the secondary (delayed) neuronal death following neuronal injury, including ischemia, traumatic brain injury (TBI) and epilepsy. The coupling of neurons by gap junctions (electrical synapses) increases during neuronal injury. In a recent study with the use of in vivo and in vitro models of cortical ischemia in mice, we have demonstrated that the ischemic increase in neuronal gap junction coupling is regulated by glutamate via group II metabotropic glutamate receptors (mGluR). Specifically, we found that activation of group II mGluRs increases background levels of neuronal gap junction coupling and expression of connexin 36 (Cx36; neuronal gap junction protein), whereas inactivation of group II mGluRs prevents the ischemia-mediated increases in the coupling and Cx36 expression. Using the analysis of neuronal death, we also established that inactivation of group II mGluRs or genetic elimination of Cx36 both dramatically reduce ischemic neuronal death in vitro and in vivo. Similar results were obtained using in vitro models of TBI and epilepsy. Our study demonstrated that mechanisms for the injury-mediated increase in neuronal gap junction coupling are part of the mechanisms for glutamate-dependent neuronal death.
Gap junctions (that are morphological correlates of electrical synapses) include intercellular channels between two neighboring cells that allow direct diffusion of ions and small molecules.Citation1 The channels are made of proteins known as connexins, which are encoded in mammals by a family of 21 connexin genes.Citation2 Connexin 36 (Cx36) is the main neuronal connexin.Citation3,Citation4 In the mammalian central nervous system (CNS), the coupling of neurons by gap junctions and the expression of Cx36 transiently increase (usually during the first two postnatal weeks) and play a role in a number of developmental events, including the regulation of neuronal death.Citation5 The coupling and expression of Cx36 then decrease,Citation6 but increase in the adult CNS following neuronal injury such as ischemia,Citation7,Citation8 spinal cord and traumatic brain injury (TBI)Citation9,Citation10 and epilepsy.Citation11,Citation12
Recently, we characterized the mechanisms that are responsible for the injury-mediated increase in neuronal gap junction coupling and the role of these mechanisms in injury-induced neuronal death.Citation13 We used oxygen-glucose deprivation (OGD) in mouse mature neuronal somatosensory cortical cultures as a model of cortical ischemia in vitro. We also used photothrombotic focal ischemia in the somatosensory cortex of adult mice as a model of ischemia in vivo. Using electrotonic coupling and western blots, we observed increases in neuronal gap junction coupling and expression of Cx36 two hours after ischemia. We established that group II metabotropic glutamate receptors (mGluR) play a role in these increases. Specifically, we showed that activation of group II mGluRs increases background levels of neuronal gap junction coupling and Cx36 expression and inactivation of group II mGluRs prevents the ischemic increases in the coupling and Cx36. We also established that the regulation by group II mGluRs is via cAMP/PKA-dependent signaling pathways (that are negatively coupled to the group II mGluRsCitation14). Further, we showed that the regulation of neuronal gap junctions likely is via action potential-dependent synaptic release of glutamate and post-transcriptional control of Cx36 expression. We also demonstrated that other neurotransmitter receptors are not involved directly in these regulatory mechanisms, including N-methyl-D-aspartate receptors (NMDAR), AMPA receptors, group I mGluRs, group III mGluRs, GABAA receptors and GABAB receptors. Moreover, activation and inactivation of group II mGluRs did not affect the expression of Cx43 (a putative glial cell connexin whose levels also increase following ischemia), suggesting that group II mGluR-dependent mechanisms do not control ischemic changes in glial gap junctions. Finally, activation of group II mGluRs did not induce coupling in Cx36-deficient neurons, indicating that these mechanisms are exclusive for regulation of Cx36.
Previously we demonstrated that pharmacological blockade of neuronal gap junctions and Cx36 knockout both prevent NMDAR-mediated excitotoxicity in vivoCitation15 and in vitroCitation16 and dramatically reduce neuronal death caused by focal cerebral ischemia in adult mice.Citation15 Therefore, in our most recent study, we tested whether mechanisms regulating ischemic increase in neuronal gap junction coupling also regulate ischemic neuronal death.Citation13 The amount of neuronal death was measured using methyl thiazolyl tetrazolium assay in neuronal cultures and Fluoro-Jade B staining in brain sections. We found that inactivation of group II mGluRs, that prevents the ischemic increase in neuronal gap junction coupling and Cx36 expression, also dramatically reduces ischemia-mediated neuronal death in vivo and in vitro. Similar results were obtained using three other in vitro injury models: hypo-osmotic shock, as a model of cytotoxic and osmotic edemas that occur during stroke and TBICitation17; hydrostatic pressure injury, that represents mechanical aspects of TBICitation18; and administration of 4-aminopyridine, as a model of epileptic seizures.Citation19 From these observations, we concluded that group II mGluRs control the injury-mediated increase in neuronal gap junction coupling and, via regulation of neuronal gap junctions, they also control death/survival mechanisms in injured neurons.Citation13
It has been suggested previouslyCitation20-Citation22 that one of the most critical factors, responsible for the secondary (delayed) neuronal death is excessive release of glutamate from injured cells that causes glutamate-dependent excitotoxicity. The excitotoxic mechanisms of glutamate are well-characterized and include hyperactivation of glutamate receptors (primarily NMDARs), massive influx of Ca2+ ions and overactivation of Ca2+-dependent signaling pathways that eventually causes death of neuronsCitation20-Citation22 (). As such, the development of NMDAR antagonists for the purposes of neuroprotection is a rational approach for the treatment of neurological disorders. Indeed, a number of NMDAR antagonists were designed and tested in different models of neuronal injury to assess their neuroprotective effects. However, clinical trials for the majority of NMDAR antagonists have failed.Citation23
Figure 1. Glutamate-dependent excitotoxicity during neuronal injuries. (A) Traditional model of the mechanisms for glutamate-dependent excitotoxicity. (B) Novel model of the mechanisms of glutamate-dependent excitotoxicity. b1: Existing neuronal gap junctions (GJ) contribute substantially to neuronal death caused by overactivation of NMDARs. b2: New neuronal gap junctions are induced by activation of group II mGluRs (IImGluRs) and also contribute to glutamate-dependent neuronal death. ⊕, this sign indicates the increase in the receptor activity or expression of Cx36. See text for details. Figure reprinted with permission: Wang Y, Song J-H, Denisova JV, Park W-M, Fontes JD, Belousov AB. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J Neurosci 2012; 32:713-25; PMID:22238107; 10.1523/jeurosci.3872-11.2012.
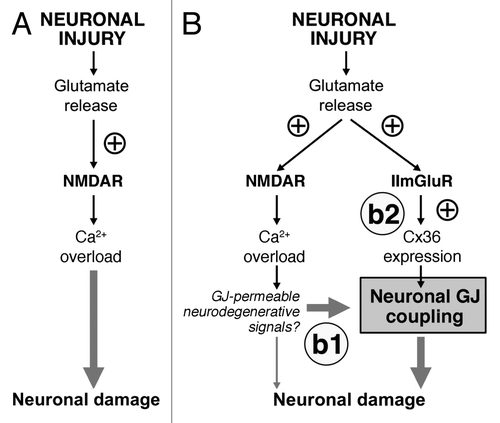
Based on our previous results and results obtained in the most recent study, we have proposed a novel model for the mechanisms of glutamate-dependent excitotoxicityCitation13 (). According to this model, during neuronal injury, the main reason for massive glutamate-dependent neuronal death is not an overactivation of NMDARs per se, but rather the expression of neuronal gap junctions. While NMDAR hyperactivity triggers neurodegenerative processes, in the absence of neuronal gap junctions this neurodegeneration is limited to a small group of neurons, which for various reasons may be especially sensitive to excitotoxicity (as we discussed earlier). However, in the presence of neuronal gap junctions, the amount of NMDAR-mediated neuronal death is multiplied and death also occurs in nearly all (or all) coupled neurons (, b1). In this respect, the background level of neuronal gap junction coupling (normally found in many mature brain regionsCitation1) is critical for NMDAR-mediated excitotoxicity to occur. In addition, an activation of group II mGluRs, caused by injury-mediated release of glutamate, induces synthesis of new neuronal gap junctions and these new gap junctions greatly enhance the extent of neuronal death (, b2).
We postulated that this is a master mechanism for neuronal death during different types of neuronal injuries, including stroke, TBI, epilepsy and presumably others. This mechanism is engaged as soon as extracellular glutamate rises to pathological levels, which triggers NMDAR excitotoxicity and initial gap junction-dependent neuronal death. The pathological mechanism then proceeds by means of the increased gap junction coupling. The coupling increases during 2–3 h post-injury, i.e., during a critical time window, where therapeutic intervention is the most successful in limiting the extent of injury-mediated cell death.
How neuronal gap junctions contribute to neuronal death is not yet understood. One possibility is by propagation between the coupled neurons of gap junction-permeable neurodegenerative signals; for example NMDAR-, AMPA receptor-, kainate receptor-, inflammation- and apoptosis-dependent signals such as Ca2+, Na+ and Ins(1,4,5)P3 (, b2). This agrees with a model of the “bystander” cell death that has been proposed previously to explain gap junction-dependent death in non-neuronal cells.Citation24,Citation25 The second possibility is the contribution via channel-independent mechanisms. This is supported by multiple lines of evidence obtained for non-neuronal connexins, suggesting that the connexins may control cell death via regulation of transcriptional programs and apoptotic pathways through direct interaction with specific factors (reviewed inCitation26). Whether the channel-dependent, or channel-independent mechanisms, or both contribute to the secondary death of neurons should be clarified in the future.
In conclusion, our study provided new mechanisms for excitotoxicity beyond ionotropic glutamate receptors that include neuronal gap junctions as a critical part of these mechanisms. The study also suggested that neuronal gap junctions should be considered as an important therapeutic target for the development of new neuroprotective agents.
| Abbreviations: | ||
| CNS | = | central nervous system |
| Cx36 | = | connexin 36 |
| mGluR | = | metabotropic glutamate receptor |
| NMDAR | = | N-methyl-D-aspartate receptor |
| OGD | = | oxygen-glucose deprivation |
| TBI | = | traumatic brain injury |
Acknowledgments
I thank the contributors to this study: Dr Yongfu Wang, Ji-Hoon Song, Janna V. Denisova, Dr Won-Mee Park, and Dr Joseph D. Fontes. This research was supported by NIH (R01 NS064256).
References
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron 2004; 41:495 - 511; http://dx.doi.org/10.1016/S0896-6273(04)00043-1; PMID: 14980200
- Söhl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci 2005; 6:191 - 200; http://dx.doi.org/10.1038/nrn1627; PMID: 15738956
- Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, et al. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A 2000; 97:7573 - 8; http://dx.doi.org/10.1073/pnas.97.13.7573; PMID: 10861019
- Belluardo N, Mudò G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, et al. Expression of connexin36 in the adult and developing rat brain. Brain Res 2000; 865:121 - 38; http://dx.doi.org/10.1016/S0006-8993(00)02300-3; PMID: 10814742
- Park W-M, Wang Y, Park S, Denisova JV, Fontes JD, Belousov AB. Interplay of chemical neurotransmitters regulates developmental increase in electrical synapses. J Neurosci 2011; 31:5909 - 20; http://dx.doi.org/10.1523/JNEUROSCI.6787-10.2011; PMID: 21508216
- Arumugam H, Liu X, Colombo PJ, Corriveau RA, Belousov AB. NMDA receptors regulate developmental gap junction uncoupling via CREB signaling. Nat Neurosci 2005; 8:1720 - 6; http://dx.doi.org/10.1038/nn1588; PMID: 16299502
- de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, et al. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke 2005; 36:2232 - 7; http://dx.doi.org/10.1161/01.STR.0000182239.75969.d8; PMID: 16179575
- Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, et al. Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci 2001; 21:7534 - 42; PMID: 11567043
- Chang Q, Pereda A, Pinter MJ, Balice-Gordon RJ. Nerve injury induces gap junctional coupling among axotomized adult motor neurons. J Neurosci 2000; 20:674 - 84; PMID: 10632597
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci 2002; 22:644 - 53; PMID: 11826094
- Gajda Z, Gyengési E, Hermesz E, Ali KS, Szente M. Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia 2003; 44:1596 - 600; http://dx.doi.org/10.1111/j.0013-9580.2003.25803.x; PMID: 14636335
- Samoilova M, Li J, Pelletier MR, Wentlandt K, Adamchik Y, Naus CC, et al. Epileptiform activity in hippocampal slice cultures exposed chronically to bicuculline: increased gap junctional function and expression. J Neurochem 2003; 86:687 - 99; http://dx.doi.org/10.1046/j.1471-4159.2003.01893.x; PMID: 12859682
- Wang Y, Song J-H, Denisova JV, Park W-M, Fontes JD, Belousov AB. Neuronal gap junction coupling is regulated by glutamate and plays critical role in cell death during neuronal injury. J Neurosci 2012; 32:713 - 25; http://dx.doi.org/10.1523/JNEUROSCI.3872-11.2012; PMID: 22238107
- Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci 2005; 6:787 - 98; http://dx.doi.org/10.1038/nrn1763; PMID: 16276355
- Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA receptor-mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol 2010; 104:3551 - 6; http://dx.doi.org/10.1152/jn.00656.2010; PMID: 20943940
- de Rivero Vaccari JC, Corriveau RA, Belousov AB. Gap junctions are required for NMDA receptor dependent cell death in developing neurons. J Neurophysiol 2007; 98:2878 - 86; http://dx.doi.org/10.1152/jn.00362.2007; PMID: 17855590
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience 2004; 129:1021 - 9; http://dx.doi.org/10.1016/j.neuroscience.2004.06.046; PMID: 15561417
- Morrison B 3rd, Saatman KE, Meaney DF, McIntosh TK. In vitro central nervous system models of mechanically induced trauma: a review. J Neurotrauma 1998; 15:911 - 28; http://dx.doi.org/10.1089/neu.1998.15.911; PMID: 9840765
- Wong M, Yamada KA. Developmental characteristics of epileptiform activity in immature rat neocortex: a comparison of four in vitro seizure models. Brain Res Dev Brain Res 2001; 128:113 - 20; http://dx.doi.org/10.1016/S0165-3806(01)00149-3; PMID: 11412897
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 2004; 61:657 - 68; http://dx.doi.org/10.1007/s00018-003-3319-x; PMID: 15052409
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988; 1:623 - 34; http://dx.doi.org/10.1016/0896-6273(88)90162-6; PMID: 2908446
- Hazell AS. Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem Int 2007; 50:941 - 53; http://dx.doi.org/10.1016/j.neuint.2007.04.026; PMID: 17576023
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury?. Lancet Neurol 2002; 1:383 - 6; http://dx.doi.org/10.1016/S1474-4422(02)00164-3; PMID: 12849400
- Cusato K, Ripps H, Zakevicius J, Spray DC. Gap junctions remain open during cytochrome c-induced cell death: relationship of conductance to ‘bystander’ cell killing. Cell Death Differ 2006; 13:1707 - 14; http://dx.doi.org/10.1038/sj.cdd.4401876; PMID: 16485029
- Peixoto PM, Ryu SY, Pruzansky DP, Kuriakose M, Gilmore A, Kinnally KW. Mitochondrial apoptosis is amplified through gap junctions. Biochem Biophys Res Commun 2009; 390:38 - 43; http://dx.doi.org/10.1016/j.bbrc.2009.09.054; PMID: 19766591
- Decrock E, Vinken M, De Vuyst E, Krysko DV, D’Herde K, Vanhaecke T, et al. Connexin-related signaling in cell death: to live or let die?. Cell Death Differ 2009; 16:524 - 36; http://dx.doi.org/10.1038/cdd.2008.196; PMID: 19197295