Abstract
Changes in intracellular calcium regulate countless biological processes. In arterial smooth muscle, voltage-dependent L-type calcium channels are major conduits for calcium entry with the primary function being determination of arterial diameter. Similarly, changes in intracellular redox status, either discrete controlled changes or global pathological perturbations, are also critical determinants of cell function. We recently reported that in arterial smooth muscle cells, local generation of hydrogen peroxide leads to colocalized calcium entry through L-type calcium channels. Here we extend our investigation into mechanisms linking hydrogen peroxide to calcium influx through L-type calcium channels by focusing on the role of protein kinase C (PKC). Our data indicate that stimulation of L-type calcium channels by hydrogen peroxide requires oxidant-dependent increases in PKC catalytic activity. This effect is independent of classical cofactor-dependent activation of PKC by diacylglycerol. These data provide additional experimental evidence supporting the concept of oxidative stimulation of L-type calcium channels.
Introduction
Voltage-dependent Cav1.2 L-type Ca2+ channels are the primary point of Ca2+ entry in mammalian arterial smooth muscle. Therefore, changes in the open probably of L-type Ca2+ channels in arterial smooth muscle cells in response to vasoconstrictors or vasodilators correlates with changes arterial diameter. Our group and others have used total internal reflection fluorescence (TIRF) microscopy to investigate mechanisms governing L-type Ca2+ channel function with high temporal and spatial resolution.Citation1-Citation6 These studies revealed that L-type Ca2+ channel functionality is heterogeneously dispersed throughout the smooth muscle cell sarcolemma as a consequence of spatially limited distributions of key regulatory molecules and subcellular processes.
Recently, our group reported that the vasoconstrictor angiotensin II (Ang II) leads to punctate generation of reactive oxygen species (ROS) by NADPH oxidase.Citation1,Citation3 This increase in local ROS production, hydrogen peroxide (H2O2) to be more precise, in turn promotes discrete colocalized Ca2+ influx events via protein kinase C (PKC)-dependent activation of L-type Ca2+ channels. Our observations suggest that oxidative activation of PKC is involved with ROS-dependent stimulation of L-type Ca2+ channels in arterial smooth muscle cells.Citation1,Citation3 Indeed, the PKC inhibitor Gö6976 prevented oxidant-dependent stimulation of L-type Ca2+ channels by exogenous ROS produced by xanthine oxidase.Citation1
Here we expand on this topic by furthering our investigation into the mechanisms underlying oxidative activation of L-type Ca2+ channels by PKC. Our data indicate that H2O2, as with ROS generated enzymatically by xanthine oxidase (which includes superoxide and H2O2) stimulates L-type Ca2+ channels in a PKC-sensitive manner. Consistent with the importance of oxidative activation of PKC, as opposed to classical activation mechanisms such as diacylglycerol (DAG), we found that inhibition of PKC activation by DAG had no effect on Ang II stimulation of L-type Ca2+ channels. In the context of our prior work,Citation1,Citation3 these data indicate that oxidant-dependent activation of arterial smooth muscle L-type Ca2+ channels by H2O2 following Ang II exposure involves oxidative activation of PKC.
Results
For our hypothesis that oxidative activation of PKC is necessary for H2O2-dependent stimulation of L-type Ca2+ channels to be valid, following oxidant exposure, inhibition of cofactor-dependent activation of PKC (i.e., via DAG) should be without effect while inhibition of PKC catalytic activity should prevent channel stimulation.
To begin, we tested the hypothesis that inhibiting the interaction between DAG and PKC (i.e., cofactor-dependent activation of PKC) would not impede oxidant-dependent stimulation of arterial smooth muscle L-type Ca2+ channels. We recorded L-type Ca2+ channel activity optically using a combination of voltage-clamp electrophysiology and TIRF microscopy as previously described.Citation1,Citation3 L-type Ca2+ channel activity was quantified in two ways: (1) the number of active L-type channel Ca2+ influx sites per µm2 (Ca2+ sparklet site density) and (2) the activity of the L-type Ca2+ channels at these sites as determined by their calculated nPs values (n is the number of quantal levels observed and Ps is the probability that the site is active).Citation5
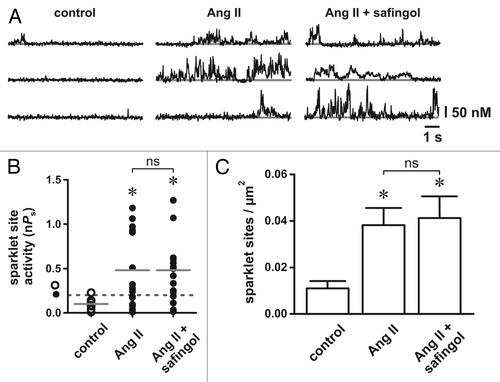
For these experiments we generated endogenous ROS by activating NADPH oxidase with Ang II.Citation7 We demonstrated previously that ROS, specifically H2O2, is necessary for oxidant-dependent stimulation of L-type Ca2+ channels in arterial smooth muscle cells.Citation3 In agreement with earlier findings,Citation1,Citation3,Citation8,Citation9 Ang II (100 nM) increased L-type Ca2+ channel activity in isolated arterial smooth muscle cells (see ). Ang II increased the density of L-type channel Ca2+ sparklet sites as well as the activity (nPs) of those sites (p < 0.05, n = 5 cells). To test if oxidative stimulation of L-type Ca2+ channels requires DAG-dependent activation of PKC we repeated our Ang II experiments in the presence of the PKC inhibitor safingol, which inhibits PKC activation by competitively interacting with the regulatory DAG/phorbol binding domain of the kinase.Citation10 Interestingly, we found that safingol (50 µM) had no effect on Ang II-dependent stimulation of L-type Ca2+ channels. As noted above, the PKC catalytic site inhibitor Gö6976 abolished stimulation of L-type Ca2+ channels by exogenous ROS generated by xanthine oxidase.Citation1 From these accumulated data we conclude that oxidative stimulation of L-type Ca2+ channels does not require DAG-dependent activation of PKC. Rather, we suggest that during increased oxidative stress, activation of PKC, which is necessary for stimulation of L-type Ca2+ channels, could occur via an oxidant-dependent mechanism.Citation11
Figure 1. Inhibition of diacylglycerol-protein kinase C interactions with safingol does not prevent angiotensin II-dependent stimulation of arterial smooth muscle L-type Ca2+ channels. (A) Representative traces showing the time course of Ca2+ influx under control conditions (left), in the presence of Ang II (100 nM; middle) and in the presence of Ang II plus safingol (10 µM; right). (B) Plot of L-type Ca2+ channel sparklet site activities (nPs) under control conditions, in the presence of Ang II and in the presence of Ang II plus safingol (n = 5 cells each). The solid gray lines are the arithmetic means of each group and the dashed line marks the threshold for high-activity Ca2+ sparklet sites (nPs ≥ 0.2). (C) Plot of the mean ± SEM L-type Ca2+ channel sparklet site densities (Ca2+ sparklet sites/µm2) under control conditions, in the presence of Ang II and in the presence of Ang II plus safingol (n = 5 cells each). * p < 0.05
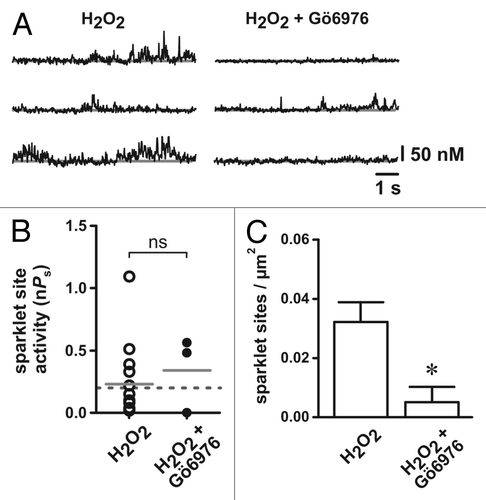
Next, we examined if H2O2-dependent stimulation of arterial smooth muscle L-type Ca2+ channels requires PKC catalytic activity by testing the effect of the PKC catalytic site inhibitor Gö6976 (100 nM) on H2O2-dependent activation of L-type Ca2+ channels. Consistent with our previous observations,Citation3 in the absence of Gö6976, H2O2 (100 µM) produced robust L-type Ca2+ channel activity (see ). In contrast, in the presence of Gö6976, H2O2 had minimal effect on L-type Ca2+ channel function. Specifically, Gö6976 abolished the increase in the number of active L-type Ca2+ channel sites observed following H2O2 exposure under control conditions (p < 0.05, n = 5 cells). Surprisingly, the activity of L-type Ca2+ channel that were observed (i.e., nPs) following H2O2 were not statistically different in the absence or presence of Gö6976 (p > 0.05, n = 5 cells). Note, however, that the number of L-type channel Ca2+ influx events observed was reduced ≈4-fold from 17 events under control conditions to only four events in the presence Gö6976 (n = 5 cells). Thus, we conclude that stimulation of L-type Ca2+ channels by H2O2 requires catalytic PKC activity.
Figure 2. Inhibition of protein kinase C catalytic activity with Gö6976 prevents H2O2-dependent stimulation of arterial smooth muscle L-type Ca2+ channels. (A) Representative traces showing the time course of Ca2+ influx in the presence of H2O2 (100 µM; left) and in the presence of H2O2 plus Gö6976 (100 nM; right). (B) Plot of L-type Ca2+ channel sparklet site activities (nPs) in the presence of H2O2 and in the presence of H2O2 plus Gö6976. (n = 5 cells each). The solid gray lines are the arithmetic means of each group and the dashed line marks the threshold for high-activity Ca2+ sparklet sites (nPs ≥ 0.2). (C) Plot of the mean ± SEM L-type Ca2+ channel sparklet site densities (Ca2+ sparklet sites/µm2) in the presence of H2O2 and in the presence of H2O2 plus Gö6976. (n = 5 cells each). * p < 0.05
Discussion
Here we continued our characterization of the role of PKC in the oxidative stimulation of arterial smooth muscle L-type Ca2+ channels. We observed that: (1) Inhibition of the interaction between DAG and PKC did not prevent Ang II from stimulating L-type Ca2+ channels, which we had previously shown to be oxidant-dependent;Citation1,Citation3 and (2) stimulation of L-type Ca2+ channels by H2O2 was abolished by inhibition of PKC catalytic activity. From these data and our published workCitation1,Citation3 we conclude that stimulation of arterial smooth muscle L-type Ca2+ channels by H2O2 involves oxidative activation of PKC.
Previous studies have shown that ROS increase the activity of PKC isoforms (such as PKCα) by oxidizing reactive cysteine residues in the kinase regulatory domain leading to constitutive cofactor-independent activity.Citation11-Citation13 Conversely, oxidation of cysteine residues in the catalytic domain leads to enzyme inactivation. Importantly, oxidative modification of the PKC regulatory and enzymatic domains is concentration dependent: Cysteine residues in the regulatory domain are more sensitive to oxidative modification than those in the catalytic domain.Citation11 As a result, limited exposure to mild oxidative conditions increases kinase activity while prolonged pathological oxidative insults promote inhibition.
We have shown that Ang II produces punctate elevations in ROS production in isolated arterial myocytes.Citation1,Citation3 Our results presented here therefore suggest a potential mechanistic basis for differential activation of discrete functional pools of PKC in arterial smooth muscle cells: PKC molecules in close proximity to sites of localized ROS generation would be subject to oxidative activation while those distal to these sites would not. Thus, targeting of PKC to specific subcellular sites (e.g., through interacting with the scaffolding protein AKAP150)Citation8 could result in initiation of oxidant-dependent and independent PKC signaling cascades with different physiological outcomes.
To conclude, our data indicate that local oxidative activation of PKC and subsequent stimulation of adjacent L-type Ca2+ channels gives rise to coordinated sites of ROS generation and Ca2+ entry. We suggest that oxidative regulation of arterial smooth muscle PKC represents a critical intracellular signaling nexus where perturbations in oxidative status translate into changes in arterial smooth muscle function via regulation of L-type Ca2+ channel activity. Our data clearly indicate that local ROS production stimulates colocalized Ca2+ influx through L-type channels.Citation1,Citation3 Interestingly, Ca2+ is known to stimulate NADPH oxidase activity via PKC.Citation14 Thus, it is reasonable to propose that colocalized ROS and Ca2+ microdomains may form a reciprocal coupling mechanism leading to sustained ROS generation and Ca2+ influx via NADPH oxidase and L-type Ca2+ channels, respectively. Future studies should address this intriguing hypothesis.
Materials and Methods
Isolation of rat cerebral arterial myocytes
Adult male Sprague-Dawley rats (Harlan) were euthanized with sodium pentobarbital (200 mg/kg intraperitoneally; Fort Dodge Animal Health) in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of Colorado State University. Isolated smooth muscle cells were prepared from basilar and cerebral arteries. Arteries were removed, cleaned and placed in ice-cold Ca2+-free buffer containing (in mM): 140 NaCl, 5 KCl, 2 MgCl2, 10 glucose and 10 HEPES (adjusted to pH 7.4 with NaOH). Arteries were incubated for 15 min at 37°C in Ca2+-free buffer supplemented with papain (10 U/mL; Worthington Biochemical) and dithiothreitol (1 mg/mL) followed by a second incubation (15 min at 37°C) in Ca2+-free buffer supplemented with collagenase (300 U/mL, Type II, Worthington Biochemical). Arteries were then washed with and placed in Ca2+-free buffer and kept on ice for 30 min after which trituration with a fire-polished Pasteur pipette was used to create a cell suspension; cells were used within 6 h of dispersion.
Electrophysiology and total internal reflection fluorescence (TIRF) microscopy
Freshly prepared smooth muscle cell suspensions were pipetted into a glass bottomed recording chamber and the cells were allowed to adhere for 20 min. Membrane potential was controlled with an Axopatch 200B amplifier (Molecular Devices). For our Ca2+ imaging experiments, we used the conventional dialyzed whole-cell patch-clamp technique. During these experiments cells were superfused with a solution containing (in mM): 120 NMDG+, 5 CsCl, 1 MgCl2, 10, glucose, 10 HEPES and 20 CaCl2 (adjusted to pH 7.4 with HCl). Pipettes were filled with a solution composed of (in mM): 87 Cs-aspartate, 20 CsCl, 1 MgCl2, 5 MgATP, 0.1 Na2GTP, 1 NADPH, 10 HEPES, 10 EGTA and 0.2 fluo-5F (adjusted to pH 7.2 with CsOH). All experiments were performed at room temperature (22–25°C).
Ca2+ influx through L-type channels was visualized with a TILL Photonics through-the-lens TIRF system built around an inverted Olympus IX-71 microscope using a 100X (numerical aperture = 1.45) TIRF oil-immersion objective and an Andor iXON EMCCD camera (Andor Technology). To monitor Ca2+ influx, myocytes were loaded with the Ca2+ indicator fluo-5F (200 µM; pentapotassium salt; Invitrogen) and an excess of EGTA (10 mM) via the patch pipette. Excitation of fluo-5F was achieved with a 491 nm laser and excitation and emission light was separated with appropriate filters. Ca2+ influx was recorded at 50 Hz at a holding potential of -70 mV and elevated external [Ca2+] (20 mM) to facilitate the detection of events and provide fluorescent signals of sufficient amplitudeCitation4 to permit quantal analysis. All experiments were allowed to progress between 5 and 10 min and only recordings with stable GΩ seals were analyzed.
L-type Ca2+ channel sparklet analysis
Background-subtracted fluo-5F fluorescence signals were converted to [Ca2+]Citation1,Citation4,Citation15 using the equation
where F is fluorescence, Fmax is the fluorescence intensity of fluo-5F in the presence of saturating free Ca2+, Fmin is the fluorescence intensity of fluo-5F in a solution where [Ca2+] is 0, Kd is the dissociation constant of fluo-5F and Rf is the Fmax/Fmin of fluo-5F. Kd and Rf values for fluo-5F were determined in vitro and Fmax was determined at the conclusion of each experiment with ionomycin (10 µM). Fluo-5F fluorescence images were analyzed with custom software.Citation4 For an elevation in [Ca2+]i to be considered an L-type Ca2+ channel sparklet event, a grid of 3 x 3 contiguous pixels had to have a [Ca2+]i amplitude equal to or larger than the mean basal [Ca2+]i plus three times its standard deviation.
L-type Ca2+ channel sparklet activity was determinedCitation1,Citation4,Citation5 by calculating the nPs of each site, where n is the number of quantal levels detected, and Ps is the probability that the site is active. nPs values were obtained using pCLAMP 10.0 (Molecular Devices) on imported [Ca2+]i time course records. L-type Ca2+ channel sparklet activity was quantified using an initial unitary [Ca2+]i elevation of 38 nM as determined experimentally.Citation5 Consistent with previous reports,Citation1,Citation2,Citation4,Citation5 L-type Ca2+ channel sparklet activity was bimodally distributed with sites of low activity (nPs between 0 and 0.2) and high activity (nPs greater than 0.2). Active L-type Ca2+ channel densities (Ca2+ sparklet sites per µm2) were calculated by dividing the number of active sites by the area of cell membrane visible in the TIRF images.
Chemicals and statistics
All chemicals were from Sigma unless stated otherwise. Normally distributed data are presented as the mean ± standard error of the mean (SEM). Two-sample comparisons of these data were performed using either a paired or unpaired (as appropriate) two-tailed Student’s t test and comparisons between more than two groups were performed using a one-way ANOVA with Tukey’s multiple comparison post-test. L-type Ca2+ channel sparklet activity (i.e., nPs) data sets were bimodally distributed, thus two-sample comparisons of nPs data were examined with the non-parametric Wilcoxon matched pairs test (two-tailed) and comparisons between more than two groups were performed using the non-parametric Friedman test with Dunn’s multiple comparison post-test. Arithmetic means of nPs data sets are indicated in the figures (solid gray horizontal lines) for non-statistical visual purposes and dashed gray lines mark the threshold for high-activity Ca2+ sparklet sites (nPs ≥ 0.2).Citation1,Citation4,Citation5 P values less than 0.05 were considered significant and asterisks (*) used in the figures indicate a significant difference between groups.
| Abbreviations: | ||
| Ang II | = | angiotensin II |
| [Ca2+]i | = | internal Ca2+ concentration |
| DAG | = | diacylglycerol |
| H2O2 | = | hydrogen peroxide |
| PKC | = | protein kinase C |
| ROS | = | reactive oxygen species |
| TIRF | = | total internal reflection fluorescence |
Acknowledgments
This work was supported by grants from the Pew Charitable Trusts and the Colorado State University College Research Council (to G.C.A.). We thank Madeline M. Frey and Adriana M. Fresquez for technical assistance.
References
- Amberg GC, Earley S, Glapa SA. Local regulation of arterial L-type calcium channels by reactive oxygen species. Circ Res 2010; 107:1002 - 10; http://dx.doi.org/10.1161/CIRCRESAHA.110.217018; PMID: 20798361
- Amberg GC, Navedo MF, Nieves-Cintrón M, Molkentin JD, Santana LF. Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J Physiol 2007; 579:187 - 201; http://dx.doi.org/10.1113/jphysiol.2006.124420; PMID: 17158168
- Chaplin NL, Amberg GC. Hydrogen peroxide mediates oxidant-dependent stimulation of arterial smooth muscle L-type calcium channels. Am J Physiol Cell Physiol 2012; 302:C1382 - 93; http://dx.doi.org/10.1152/ajpcell.00222.2011; PMID: 22322977
- Navedo MF, Amberg GC, Nieves M, Molkentin JD, Santana LF. Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle. J Gen Physiol 2006; 127:611 - 22; http://dx.doi.org/10.1085/jgp.200609519; PMID: 16702354
- Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active L-type Ca2+ channels. Proc Natl Acad Sci U S A 2005; 102:11112 - 7; http://dx.doi.org/10.1073/pnas.0500360102; PMID: 16040810
- Navedo MF, Amberg GC, Westenbroek RE, Sinnegger-Brauns MJ, Catterall WA, Striessnig J, et al. Ca(v)1.3 channels produce persistent calcium sparklets, but Ca(v)1.2 channels are responsible for sparklets in mouse arterial smooth muscle. Am J Physiol Heart Circ Physiol 2007; 293:H1359 - 70; http://dx.doi.org/10.1152/ajpheart.00450.2007; PMID: 17526649
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994; 74:1141 - 8; http://dx.doi.org/10.1161/01.RES.74.6.1141; PMID: 8187280
- Navedo MF, Nieves-Cintrón M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, et al. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res 2008; 102:e1 - 11; http://dx.doi.org/10.1161/CIRCRESAHA.107.167809; PMID: 18174462
- Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci U S A 2008; 105:15623 - 8; http://dx.doi.org/10.1073/pnas.0808759105; PMID: 18832165
- Hannun YA, Loomis CR, Merrill AH Jr., Bell RM. Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J Biol Chem 1986; 261:12604 - 9; PMID: 3462188
- Gopalakrishna R, Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A 1989; 86:6758 - 62; http://dx.doi.org/10.1073/pnas.86.17.6758; PMID: 2505261
- Knapp LT, Klann E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J Neurosci 2002; 22:674 - 83; PMID: 11826097
- Palumbo EJ, Sweatt JD, Chen SJ, Klann E. Oxidation-induced persistent activation of protein kinase C in hippocampal homogenates. Biochem Biophys Res Commun 1992; 187:1439 - 45; http://dx.doi.org/10.1016/0006-291X(92)90463-U; PMID: 1417820
- Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 2009; 47:1239 - 53; http://dx.doi.org/10.1016/j.freeradbiomed.2009.07.023; PMID: 19628035
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J 2000; 78:2655 - 67; http://dx.doi.org/10.1016/S0006-3495(00)76809-3; PMID: 10777761