Abstract
In our recent publication, we describe the local anesthetic (LA) inhibition of the prokaryotic voltage gated sodium channel NaChBac. Despite the numerous functional and putative structural differences with the mammalian sodium channels, the data show that LA compounds effectively and reversibly inhibit NaChBac channels in a concentration range similar to resting blockade on eukaryotic Navs. In addition to current reduction, LA application accelerated channel inactivation kinetics of NaChBac which could be accounted for in a simple state-model whereby local anesthetics increase the probability of entering the inactivated state. We have further explored what state (or states) local anesthetic blockade of NaChBac could pertain to eukaryotic sodium channels, and what molecular similarities exist between these disparate channel families. Here we show that the rate of recovery from inactivation remains unaffected in the presence of local anesthetics. Further, we show that two sites that support use-dependent inhibition in eukaryotic channels, do not affect block to the same extent when mutated in NaChBac channels. The data indicate that the molecular determinants and the inherent mechanisms for LA block are likely to be divergent between bacterial and eukaryotic Navs, but future experiments will help define possible similarities.
Introduction
Voltage-gated sodium channels are key regulators of electrical signaling in the cells of nerve and muscle excitable cells.Citation1 These channels display sub-millisecond activation kinetics, and the resultant rapid influx of sodium ions drives the initiation of the action potential making them key players in maintaining electrical excitability and stability in the cells of cardiac and neuronal tissues. Seemingly modest alternations in Nav channel gating parameters can result in a myriad of pathological conditions including lethal cardiac arrhythmia, severe epilepsy and debilitating pain syndromes, conditions which can be effectively managed by therapeutics that inhibit sodium ion conductance.Citation2-Citation6
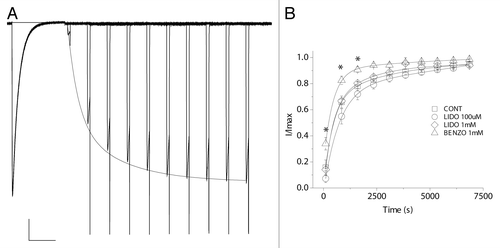
The prokaryotic family of voltage-gated sodium channels, including NaChBac from Bacillus halodurans, are homotetramers with modest sodium ion selectivity and voltage-dependent gating.Citation7-Citation9 Recent crystallographic breakthroughs with the bacterial Nav homologs NavAb and NavRh have begun to shed light on the details of sodium ion selectivity and to capture conformations which may represent functional gating states of the channel.Citation10-Citation12 The molecular determinants of NaChBac inactivation remain undescribed but could involve conformational changes in the selectivity filter, akin to slow inactivation in potassium channels,Citation13 or could be due to re-closure or “slippage” of the S6 activation gate.Citation14 In terms of pharmacology, these structures depict a large cytoplasmic cavity that could accommodate a large blocker such as a local anesthetic molecule, but one of the most intriguing aspects of these new structures is the presence of fenestrations that could, in theory, allow direct access of drugs to the inner vestibule from the membrane. While such portals can be found in the potassium channel structures of KcsACitation15 and K2P,Citation16,Citation17 their presence here could provide a molecular explanation for putative “hydrophobic pathways” that could allow for closed-state blockade of eukaryotic channels.Citation18 In our recent publication, we reported that the eukaryotic sodium channel blockers benzocaine, lidocaine and ranolazine inhibit NaChBac current with an affinity comparable to the resting block of the mammalian channels.Citation19 Sidedness of blockade by the impermeant blocker QX-314 suggested the receptor for the charged form of the blockers was cytoplasmic. Further, a novel mutation, T220A, in the pore-lining S6 segment, was shown to abolish channel inactivation and was employed to reveal that blocking kinetics were fast or reached equilibrium in the closed state, producing an instantaneous reduction in current upon channel opening. We also observed a concentration-dependent increase in the rate of inactivation in WT channels upon drug exposure, which may be explained by a simple kinetic scheme whereby the drug binding promotes entry into the inactivated state. We now show that the rate of recovery from inactivation in the presence of lidocaine was not affected (). This observation is in contrast to the significant stabilization of the fast-inactivated state which can manifest as a slowing of recovery from fast inactivation seen in eukaryotic Navs in the presence of local anesthetic compounds.Citation20 Thus, it is likely that NaChBac adopts an alternative blocking mode, or mechanism, compared with eukaryotic Nav channels which could underlie differences in drug effects on inactivation properties.
Figure 1. The rate of recovery from inactivation is unaffected by lidocaine. Channels were held at holding potential of -120 mV and activated by 750 ms of depolarizing pulses to -20 mV every 20 sec. (A) A representative family of current traces of WT NaChBac, showing the step-wise recovery from inactivation. Scale bar represents 1 sec and 2 nA. (B) Normalized peak currents are plotted as a function of recovery time interval. The rate of recovery from inactivation is significantly increased in the presence of 1 mM benzocaine, where asterisks indicate (p < 0.05).
The Putative LA-Binding Sites Are Not Conserved in NaChBac
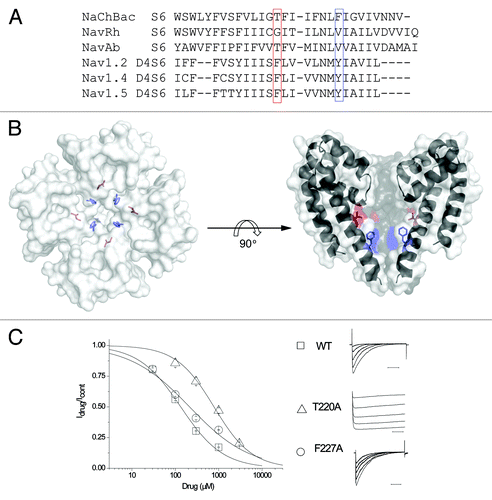
Do bacterial channels provide a long-sought solution for structure-based drug design in sodium channels? Resting blockade and use-dependent inhibition of eukaryotic sodium channels each rely on distinct molecular interactions with different amino acids in the local anesthetic receptor. We aimed to determine which residues support inhibition of NaChBac channels, and if these sites corresponded to those essential for use-dependent inhibition of eukaryotic channels. The sequence comparison between the S6 pore-lining segments of NaChBac and those from eukaryotic sodium channels are shown in , illustrating the partial conservation of residues implicated in supporting use-dependent inhibition in eukaryotic Nav’s. Point mutations of F1764 and Y1771 in Nav1.2, and the corresponding residues in all eukaryotic Nav isoforms, ameliorate use-dependent inhibition by local anesthetic, anti-arrhythmic and anti-epileptic compounds.Citation21,Citation22 It is noteworthy that the aromatic, highlighted by the red box in , which supports lidocaine use-dependent block in eukaryotic channels through a cation-pi interaction,Citation23,Citation24 is entirely unconserved in bacterial channels. To gain more information on the nature of drug block in NaChBac, we examined lidocaine inhibition in T220A and F227A mutant channels (). Surprisingly, alanine mutation of the two residues T220 and F227 in NaChBac showed little variation in lidocaine inhibition IC50s, with ~6-fold and ~2-fold loss of affinity, respectively (). For comparison, the homologous mutations in Nav1.2 nearly abolish or severely impair use-dependent inhibition for F1764A or Y1771A, respectively.Citation21 Attempts to study the “rescued” pharmacology T220F channel were not possible as this mutant displayed a non-expressing phenotype. Ongoing and future mutagenic studies will serve to further define the determinants of bacterial sodium channel inhibition, and if these coordinates are shared with sites previously identified to support resting block of eukaryotic channels.
Figure 2. The canonical local anesthetic-binding sites are not conserved in NaChBac. (A) The sequence alignment of S6 of bacterial and eukaryotic S6 segments, highlighting two sites implicated in use-dependent inhibition of eukaryotic channels. (B) Top-down view of the highlighted residues shown on the right. (C) A concentration response curve was generated with the mean of Idrug/Icontrol obtained from the peak currents after exposing cells to 30 μM, 100 μM, 300 μM, 1 mM and 3 mM (for T220A only) lidocaine. The IC50 for lidocaine, according to the concentration response curve, is ~135 μM for WT(), ~788 μM for T220A() and ~214 μM for F227A (). Representative current traces are obtained after depolarization from the holding potential -120 to -20 mV in the presence of various lidocaine concentrations, peak current sizes were reduced upon application of increasing lidocaine concentrations (right).
Materials and Methods
The wild-type and mutant NaChBac channels were transiently expressed via calcium phosphate transfection in HEK-293 cells for patch clamp recordings. The patch pipette contained (in mmol/L): 60 CsCl2, 13 NaCl, 80 K-L-Aspartic Acid, 11 EGTA, 1 MgCl2, 1 CaCl2, 10 HEPES (pH 7.2), the bath contained (in mmol/L) 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2 and 10 HEPES (pH 7.4). Electrode resistance was in the range of 1.0–1.5 MΩ voltage errors due to series resistance was always < 3 mV after compensation and liquid junction potentials were corrected. All experiments were performed at room temperature.
Conclusions
In addition to the inhibition of the bacterial sodium channel described here, lidocaine can rapidly and reversibly block a variety of channels and receptor types, including KATP, K2P, T-type Ca2+, hyperpolarization activated channels in addition to nicotinic acetylcholine receptors as well as eukaryotic sodium channels, suggesting that a minimal common molecular architecture is shared among these channel types.Citation25-Citation29 However, the recent appearance of bacterial sodium channel structures represents a significant step toward the long-held goal of structure-based drug design in sodium channels. The pharmacological and functional commonalities shared between bacterial and eukaryotic channels have been, thus far, limited by lack of detailed information on bacterial gating and pharmacological parameters. In this light, our data on local anesthetic inhibition of NaChBac has begun to highlight potential similarities and differences between these disparate channels.
References
- Hille B. Ion channels of excitable membranes. 3rd ed2001, Sunderland, Mass.: Sinauer. xviii, 814 p.
- Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 2009; 6:337 - 48; http://dx.doi.org/10.1038/nrcardio.2009.44; PMID: 19377496
- Roden DM, George AL Jr.. The cardiac ion channels: relevance to management of arrhythmias. Annu Rev Med 1996; 47:135 - 48; http://dx.doi.org/10.1146/annurev.med.47.1.135; PMID: 8712768
- Waxman SG, Hains BC. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci 2006; 29:207 - 15; http://dx.doi.org/10.1016/j.tins.2006.02.003; PMID: 16494954
- Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci USA 1999; 96:7635 - 9; http://dx.doi.org/10.1073/pnas.96.14.7635; PMID: 10393872
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 2005; 115:2010 - 7; http://dx.doi.org/10.1172/JCI25466; PMID: 16075041
- Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE. A prokaryotic voltage-gated sodium channel. Science 2001; 294:2372 - 5; http://dx.doi.org/10.1126/science.1065635; PMID: 11743207
- Blanchet J, Pilote S, Chahine M. Acidic residues on the voltage-sensor domain determine the activation of the NaChBac sodium channel. Biophys J 2007; 92:3513 - 23; http://dx.doi.org/10.1529/biophysj.106.090464; PMID: 17325004
- Yue L, Navarro B, Ren D, Ramos A, Clapham DE. The cation selectivity filter of the bacterial sodium channel, NaChBac. J Gen Physiol 2002; 120:845 - 53; http://dx.doi.org/10.1085/jgp.20028699; PMID: 12451053
- Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 2012; 486:130 - 4; PMID: 22678295
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012; 486:135 - 9; PMID: 22678296
- Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature 2011; 475:353 - 8; http://dx.doi.org/10.1038/nature10238; PMID: 21743477
- Pavlov E, Bladen C, Winkfein R, Diao C, Dhaliwal P, French RJ. The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys J 2005; 89:232 - 42; http://dx.doi.org/10.1529/biophysj.104.056994; PMID: 15849254
- Shin KS, Maertens C, Proenza C, Rothberg BS, Yellen G. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage. Neuron 2004; 41:737 - 44; http://dx.doi.org/10.1016/S0896-6273(04)00083-2; PMID: 15003173
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998; 280:69 - 77; http://dx.doi.org/10.1126/science.280.5360.69; PMID: 9525859
- Miller AN, Long SB. Crystal structure of the human two-pore domain potassium channel K2P1. Science 2012; 335:432 - 6; http://dx.doi.org/10.1126/science.1213274; PMID: 22282804
- Brohawn SG, del Mármol J, MacKinnon R. Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 2012; 335:436 - 41; http://dx.doi.org/10.1126/science.1213808; PMID: 22282805
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol 1977; 69:497 - 515; http://dx.doi.org/10.1085/jgp.69.4.497; PMID: 300786
- Lee S, Goodchild SJ, Ahern CA. Local anesthetic inhibition of a bacterial sodium channel. J Gen Physiol 2012; 139:507 - 16; http://dx.doi.org/10.1085/jgp.201210779; PMID: 22641643
- Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol 1983; 81:613 - 42; http://dx.doi.org/10.1085/jgp.81.5.613; PMID: 6306139
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994; 265:1724 - 8; http://dx.doi.org/10.1126/science.8085162; PMID: 8085162
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA 1996; 93:9270 - 5; http://dx.doi.org/10.1073/pnas.93.17.9270; PMID: 8799190
- Pless SA, Galpin JD, Frankel A, Ahern CA. Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat Commun 2011; 2:351; http://dx.doi.org/10.1038/ncomms1351; PMID: 21673672
- Ahern CA, Eastwood AL, Dougherty DA, Horn R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ Res 2008; 102:86 - 94; http://dx.doi.org/10.1161/CIRCRESAHA.107.160663; PMID: 17967784
- Olschewski A, Bräu ME, Olschewski H, Hempelmann G, Vogel W. ATP-dependent potassium channel in rat cardiomyocytes is blocked by lidocaine. Possible impact on the antiarrhythmic action of lidocaine. Circulation 1996; 93:656 - 9; http://dx.doi.org/10.1161/01.CIR.93.4.656; PMID: 8640992
- Kindler CH, Paul M, Zou H, Liu C, Winegar BD, Gray AT, et al. Amide local anesthetics potently inhibit the human tandem pore domain background K+ channel TASK-2 (KCNK5). J Pharmacol Exp Ther 2003; 306:84 - 92; http://dx.doi.org/10.1124/jpet.103.049809; PMID: 12660311
- Alberola-Die A, Martinez-Pinna J, González-Ros JM, Ivorra I, Morales A. Multiple inhibitory actions of lidocaine on Torpedo nicotinic acetylcholine receptors transplanted to Xenopus oocytes. J Neurochem 2011; 117:1009 - 19; http://dx.doi.org/10.1111/j.1471-4159.2011.07271.x; PMID: 21480901
- Bladen C, Zamponi GW. Common mechanisms of drug interactions with sodium and T-type calcium channels. Mol Pharmacol 2012; 82:481 - 7; http://dx.doi.org/10.1124/mol.112.079715; PMID: 22695716
- Meng QT, Xia ZY, Liu J, Bayliss DA, Chen X. Local anesthetic inhibits hyperpolarization-activated cationic currents. Mol Pharmacol 2011; 79:866 - 73; http://dx.doi.org/10.1124/mol.110.070227; PMID: 21303986