Abstract
The Orai family of calcium channels includes the store-operated CRAC channels and store-independent, arachidonic acid (AA)-regulated ARC channels. Both depend on STIM1 for their activation but, whereas CRAC channel activation involves sensing the depletion of intracellular calcium stores via a luminal N terminal EF-hand of STIM1 in the endoplasmic reticulum (ER) membrane, ARC channels are exclusively activated by the pool of STIM1 that constitutively resides in the plasma membrane (PM). Here, the EF-hand is extracellular and unlikely to ever lose its bound calcium, suggesting that STIM1-dependent activation of ARC channels is very different from that of CRAC channels. We now show that attachment of the cytosolic portion of STIM1 to the inner face of the PM via an N terminal Lck-domain sequence is sufficient to enable normal AA-dependent activation of ARC channels, while failing to allow activation of store-operated CRAC channels. Introduction of a point mutation within the Lck-domain resulted in the loss of both PM localization and ARC channel activation. Reversing the orientation of the PM-anchored STIM1 C terminus via a C-terminal CAAX-box fails to support either CRAC or ARC channel activation. Finally, the Lck-anchored STIM1 C-terminal domain also enabled the exclusive activation of the ARC channels following physiological agonist addition. These data demonstrate that simple tethering of the cytosolic C-terminal domain of STIM1 to the inner face of the PM is sufficient to allow the full, normal and exclusive activation of ARC channels, and that the N-terminal regions of STIM1 (including the EF-hand domain) play no significant role in this activation.
Introduction
Orai proteins form the structural subunits of a family of voltage-independent calcium channels that includes both the store-operated calcium release-activated calcium (CRAC) channels and the store-independent arachidonic acid-regulated calcium (ARC) channels. These two channel types are biophysically similar in that they are small, highly calcium-selective conductances and both have been shown to play important roles in agonist-activated calcium entry, particularly in non-excitable cells. However, they differ in their molecular composition in that, while the functional CRAC channel is formed by a homotetrameric assembly of Orai1 proteins,Citation1-Citation3 the ARC channel is a heteropentamer comprised of three Orai1 and two Orai3 subunits.Citation4,Citation5 Critically, although both channels have been shown to depend on STIM1 for their activation, entirely distinct pools of the protein are responsible. Physiologically, CRAC channel activation relies on the STIM1 that resides in the membrane of the endoplasmic reticulum (ER).Citation6,Citation7 Here, the key initiating step in activation of the channels is the loss of calcium from a luminally located N-terminal EF-hand domain of STIM1, following depletion of calcium from the ER store. This results in conformational changes in other regions of the protein that, in turn, induce the oligomerization of the STIM1 molecules and their translocation within the ER membrane to sites close to the plasma membrane,Citation8-Citation12 where the cytosolic regions of STIM1 interact with the CRAC channels to result in their gating.Citation13,Citation14 In contrast, activation of the ARC channels exclusively depends on the pool of STIM1 that constitutively resides in the plasma membrane,Citation15 a component that typically constitutes some 15–25% of the total cellular STIM1.Citation15,Citation16 This location raises an interesting question in that the calcium-binding N-terminal EF-hand of STIM1 would lie in the extracellular medium where, given its reported Kd for calcium of ~300–600 µM,Citation17,Citation18 it is unlikely to ever lose its bound calcium under normal circumstances. Consequently, it would seem that the STIM1-dependent activation of the ARC channels is likely to display marked differences from that of the CRAC channels.
Examination of any such differences raises the problem of being able to definitively distinguish between the STIM1-dependent activation of the ARC channels and that of the co-existing CRAC channels. Selective activation of CRAC channels can be achieved by expression of STIM1 constructs that are unable to be inserted in the plasma membrane,Citation15,Citation19 but no equivalent constructs exist for the ARC channels. Consequently, the aim of the present study was to develop a system which would permit the exclusive activation of the ARC channels without any corresponding activation of the co-existing CRAC channels. In addition, we specifically sought a system that was capable of activating the endogenous channels, thereby avoiding any possible complications arising from the consequences of overexpression. As a result, we now demonstrate that the simple tethering of the cytosolic portion of STIM1 to the inner surface of the plasma membrane in the appropriate orientation is sufficient to permit the normal activation of endogenous ARC channels, while failing to induce any measureable activation of the co-existing CRAC channels, either in the presence or absence of depletion of the ER calcium stores. Critically, the activity of the ARC channels under these conditions is not constitutive, but still requires the addition of exogenous arachidonic acid, or the action of an appropriate agonist.
Results
Expression of the free C terminus of STIM1
As noted, the extracellular location of the N-terminal EF-hand of plasma membrane STIM1 would mean that it is unlikely to ever lose its bound calcium, resulting in the corresponding absence of any subsequent conformational changes in the adjacent SAM domain, suggesting that these N-terminal regions of the molecule might be redundant in the STIM1-dependent activation of the ARC channels. Moreover, existing evidence from studies on the CRAC channels indicates that the actual gating of the channel is determined by interactions between Orai1 and the cytosolic C-terminal regions of STIM1.Citation20,Citation21 Based on these findings, we first explored an approach that has been widely used in studies on activation of the CRAC channels involving the expression of a STIM1 construct consisting of just the free cytosolic regions of the molecule (i.e., lacking both the N terminal and transmembrane domains).Citation22-Citation24 For this, we modified a STIM1 C-terminal construct, comprising residues L251–K685Citation25 to incorporate silent mutations that rendered it insensitive to a STIM1 siRNA. In this way, expression of this construct in siRNA-treated cells allowed examination of its ability to activate endogenous CRAC and ARC channels while minimizing any contribution from the endogenous STIM1. We have previously shown that expression of such an siRNA-resistant STIM1 construct is able to fully rescue normal endogenous ARC channel and CRAC channel activity in HEK293 cells treated with the STIM1 siRNA.Citation15
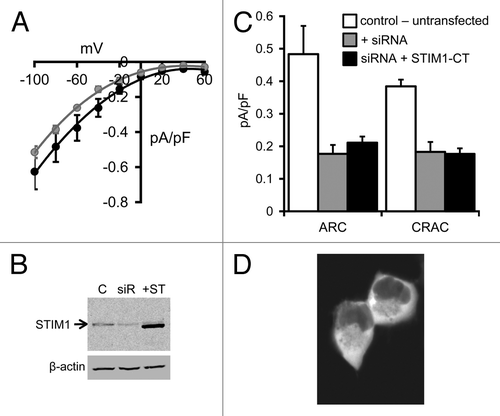
The presence of both ARC and CRAC channels in the FlpIn HEK293 cells was demonstrated by the appearance of typically small inwardly-rectifying currents following exogenous addition of arachidonic acid (8 µM), or maximal depletion of intracellular stores by inclusion of adenophostin-A (2 µM) in a Ca2+-free pipette solution, respectively (). The magnitudes of these currents are similar to those recorded in normal, untransfected HEK293 cells.Citation15 Treatment of these cells with siRNA targeting endogenous STIM1 markedly reduced measured levels of STIM1 protein as assessed by western blot using a STIM1-specific antibody (), and reduced maximal arachidonic acid (8 µM) activated ARC channel currents measured at -80 mV from a value of 0.48 ± 0.09 pA/pF (n = 7) in control (untransfected) cells, to only 0.18 ± 0.03 pA/pF (n = 5) (). Similarly, the corresponding store-operated CRAC channel currents were reduced from 0.38 ± 0.02 pA/pF (n = 7) in control cells, to 0.18 ± 0.03 pA/pF (n = 5) in the siRNA-treated cells. Confocal imaging of cells following expression of the siRNA-resistant STIM1 C-terminal (STIM1-CT) construct bearing a C-terminal eGFP tag revealed that the protein displayed a diffused fluorescence signal consistent with its anticipated distribution throughout the cytosol (). Examination of the arachidonic acid-induced currents in cells expressing the free STIM1-CT construct failed to induce the activation of significant ARC channel currents above levels seen in the siRNA-treated cells (0.21 ± 0.02 pA/pF at -80 mV, n = 5, p = 0.33). Similarly, following maximal depletion of intracellular calcium stores by use of the adenophostin-A/Ca2+-free pipette solution, measured store-operated CRAC currents in the STIM1-CT expressing cells were essentially identical to those recorded in the cells treated with STIM1 siRNA alone (0.18 ± 0.02 pA/pF at -80 mV, n = 6, p = 0.92) (). Western blot analysis of cell extracts showed that the free STIM1-CT construct was clearly expressed in the transfected cells (), so its failure to activate either endogenous ARC or CRAC channels was not due to a lack of expression. It should be noted that our finding that the expressed free cytosolic C terminus of STIM1 is unable to activate the CRAC channels is contrary to several reports indicating that such expression results in a constitutive activation of either store-operated Ca2+ entry,Citation22 or CRAC channel currents (see for example, refs. Citation24, Citation26 and Citation27). However, most of these studies involved the co-expression of the STIM1-CT along with Orai1, and, at least in HEK cells, while STIM1-CT expressed alone remains cytosolic and fails to induce any significant CRAC currents, co-expression with Orai1 results in a redistribution of the STIM1-CT to the plasma membrane, and the constitutive activation of the CRAC channels.Citation23 Moreover, studies have also indicated that such constitutive activity is highly dependent on the expression levels of these protein partners.Citation13,Citation23 Consequently, consistent with these latter reports, we conclude that expression of the free cytosolic C-terminal region of STIM1 alone, at least in HEK293 cells and at the levels employed here, not only fails to induce any detectible constitutive activation of endogenous CRAC or ARC channels, but is also unable to support any significant activation of these channels following store-depletion or addition of exogenous arachidonic acid, respectively.
Figure 1. ARC and CRAC channel currents in HEK293 cells expressing the STIM1-CT construct. (A) Mean (± SE) I/V curves for the endogenous AA-activated ARC channels (black) and store-operated CRAC channels (gray) in HEK293 cells. (B) Representative western blot showing STIM1 levels in untransfected cells (C), in the same cells following treatment with a STIM1 siRNA (siR) and in the siRNA-treated cells following expression of the siRNA-resistant STIM1-CT construct (+ST). (C) Mean (± SE) ARC currents and CRAC currents measured at -80 mV in untransfected cells (white columns), in STIM1 siRNA-treated cells (gray columns) and in siRNA-treated cells following expression of the siRNA-resistant STIM1-CT construct (black columns). (D) Representative live confocal image of cells expressing the eGFP-tagged STIM1-CT construct.
Plasma membrane targeting of the STIM1 C terminus
As already noted, a key step in the activation of the CRAC channels following calcium store depletion is the translocation of oligomers of STIM1 located in the ER membrane to sites close to the plasma membrane where the cytosolic C-terminal regions of the protein can interact with the Orai1 subunits that form the channels. We therefore considered whether a corresponding “spatial association” might also be critical for the ability of STIM1 in the plasma membrane to effectively activate the ARC channels. To examine this, we chose to try and anchor the STIM1-CT protein, in the appropriate orientation, to the inner face of the plasma membrane using a short N-terminal sequence based on the SH4 domain (Src homology domain 4) of Lck, a Src-related protein tyrosine kinase. This Lck domain comprises 10 residues (M-G-C-G-C-S-S-H-P-E), of which the first glycine is a site for the stable cotranslational linkage of the fatty acid myristate. This myristoylation then allows the further addition of palmitate to cysteine residues within the Lck domain sequence,Citation28,Citation29 resulting in the selective anchoring of the domain to the inner face of the plasma membrane. Consequently, numerous studies have demonstrated that the simple attachment of the Lck domain sequence to various proteins or peptides represents an effective way to anchor such molecules at the plasma membrane.Citation30-Citation32 We therefore modified the siRNA-resistant STIM1-CT construct by attaching the Lck domain to the N terminus of the STIM1-CT via a short, flexible, hydrophilic 7-residue linker sequenceCitation32 to give a construct designated as Lck-STIM1-C.
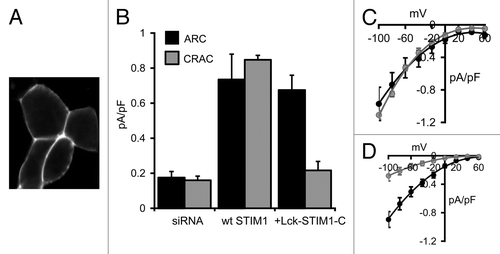
Confocal imaging of cells expressing the Lck-STIM1-C construct bearing a C-terminal eGFP tag revealed a clear constitutive association of the construct with the plasma membrane (). Similar to the data above, treatment of the cells with an siRNA targeting STIM1 reduced stored depletion-dependent CRAC channel currents and arachidonic acid-induced ARC channel currents, measured at -80 mV, to values of only 0.16 ± 0.02 pA/pF (n = 5) and 0.17 ± 0.04 pA/pF (n = 5), respectively (). As with the free cytosolic STIM1-C construct above, expression of the Lck-STIM1-C (without the C-terminal eGFP tag) in STIM1 siRNA-treated cells did not result in any significant constitutively active currents. However, addition of exogenous arachidonic acid (8 µM) resulted in the appearance of relatively large, inwardly rectifying currents (0.67 ± 0.09 pA/pF, n = 7) that were equivalent to those seen in the same siRNA-treated cells on expression of an siRNA-resistant, full-length wild-type STIM1 (0.73 ± 0.15 pA/pF, n = 4, p = 0.71) (). Clearly, the anchoring of the cytosolic STIM1 C terminus to the inner face of the plasma membrane was sufficient to allow the full restoration of normal arachidonic acid-activated ARC channel currents. In marked contrast, currents measured in the same cells following depletion of the calcium stores with the adenophostin-A/Ca2+-free pipette solution were only 0.22 ± 0.05 pA/pF (n = 6), a value markedly different from the corresponding siRNA-treated cells expressing a wild-type STIM1 construct (0.85 ± 0.03 pA/pF, n = 4) (). Indeed, CRAC channel currents in the Lck-STIM1-C-expressing cells were not significantly different from those recorded in the control siRNA-treated cells (p = 0.36). These data clearly indicate that, in marked contrast to ARC channel currents, the Lck-STIM1-C construct was unable to induce normal activation of the store-operated CRAC channels.
Figure 2. Expression of the Lck-STIM1-C construct in HEK293 cells. (A) Representative live confocal image of cells expressing the Lck-STIM1-CT construct bearing a C-terminal eGFP-tag. (B) Mean (± SE) AA-activated ARC channel currents (black) and store-operated CRAC channel currents (gray) measured at -80 mV in siRNA-treated cells (siRNA), and in the same cells following expression of an siRNA-resistant wild-type STIM1 (+wt STIM1) or the siRNA-resistant Lck-STIM1-C construct (+Lck-STIM1-C). (C) Mean (± SE) I/V curves for the ARC channels (black) and CRAC channels (gray) in the siRNA-treated cells expressing the wild-type STIM1, and (D) the corresponding I/V curves for the ARC channels and CRAC channels in the siRNA-treated cells expressing the Lck-STIM1-C construct.
Lck-STIM1-C and CRAC channels in RBL cells
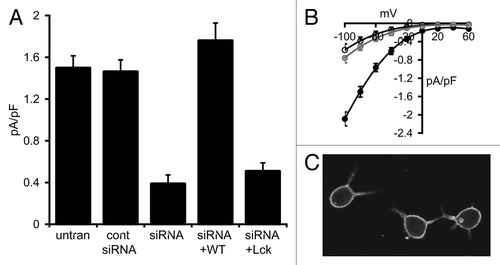
To confirm this apparent failure of the Lck-STIM1-C construct to support activation of the CRAC channels, we turned to RBL-1 cells, a cell type which has been extensively used to study CRAC channels, and whose currents are significantly larger than those typically seen in HEK cells. Correspondingly, store-operated inward CRAC currents measured at -80 mV in these cells averaged 1.50 ± 0.12 pA/pF (n = 6), approximately four times larger than those recorded in the HEK293 cells (). Following transfection with the STIM1 siRNA, store-operated CRAC channel currents in these cells were reduced by some 75% to a value of 0.39 ± 0.09 pA/pF (n = 4), while expression of a control siRNA did not significantly affect store-operated currents (1.46 ± 0.11 pA/pF, n = 4) (). As in the HEK cell experiments above, confocal microscopy of RBL cells expressing an eGFP-tagged Lck-STIM1-C construct indicated a distribution consistent with a constitutive association with the plasma membrane (). However, again as seen in the HEK cells, expression of the siRNA-resistant Lck-STIM1-C construct in siRNA-treated RBL cells failed to significantly increase store-operated currents (0.51 ± 0.08 pA/pF, n = 6, p = 0.35) (). In marked contrast, expression of an siRNA-resistant, full-length wild-type STIM1 construct in the siRNA-treated cells resulted in the full restoration of normal store-operated CRAC channels currents to a value (1.76 ± 0.17 pA/pF, n = 6) that was not significantly different from untransfected cells (p = 0.23) (). Together, these data confirm the inability of the Lck-STIM1-C construct to effectively enable the activation of the CRAC channels by store depletion.
Figure 3. Effect of the Lck-STIM1-C construct on CRAC channel activity in RBL cells. (A) Mean (± SE) store-operated CRAC channel currents measured at -80 mV in untransfected cells (untran), in cells expressing a control siRNA (cont siRNA), a STIM1 siRNA (siRNA) and in the same STIM1 siRNA-treated cells expressing an siRNA-resistant wild-type STIM1 (siRNA+WT) or an siRNA-resistant Lck-STIM1-C construct (siRNA+Lck). (B) Mean (± SE) I/V curves for CRAC channels in untransfected RBL cells (black), in STIM1 siRNA-treated cells (open circles) and in the same cells expressing the siRNA-resistant Lck-STIM1-C construct (gray circles). (C) Representative live confocal image of RBL cells expressing the Lck-STIM1-CT construct bearing a C-terminal eGFP-tag.
Molecular disruption of Lck-dependent plasma membrane targeting
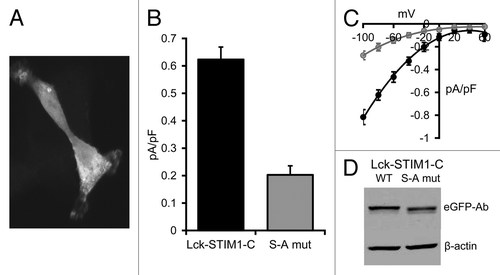
In the studies above, we assumed that the ability of the Lck-STIM1-C construct to enable the exclusive arachidonic acid-dependent activation of the ARC channels resulted from the ability of the attached Lck domain to anchor the STIM1-C protein at the inner face of the plasma membrane. To confirm this, we examined the effect of selectively eliminating such targeting using a molecular approach. Studies have shown that the introduction of a single-point mutation (S to A) at position 6 in the Lck domain sequence results in a profound reduction in the myristoylation of the initial glycine residue, and a consequent loss of specific plasma membrane targeting of the domain.Citation33 As anticipated, introduction of this S to A mutation in the Lck-STIM1-C construct resulted in the loss of selective plasma membrane targeting of the construct, as assessed by confocal imaging of an eGFP-tagged construct in HEK cells which, instead, displayed a diffused cytoplasmic fluorescence signal similar to that seen previously with the free STIM1-C construct (). Correspondingly, expression of this S to A mutant Lck-STIM1-C construct in siRNA-treated HEK cells markedly reduced arachidonic acid-activated ARC channel currents from a value of 0.62 ± 0.05 pA/pF (n = 6) at -80 mV in cells expressing the normal Lck-STIM1-C, to only 0.20 ± 0.03 pA/pF (n = 6) in the cells expressing the mutant Lck-STIM1-C (). Western blot analysis of extracts made from the transfected cells showed that both the Lck-STIM1-C and mutant Lck-STIM1C mutant were expressed at almost a similar level (). Consequently, the data indicate that the expression of the Lck-STIM1-C construct bearing the single critical S to A mutation in the Lck domain essentially eliminated the ability of the construct to support the specific activation of endogenous ARC channels.
Figure 4. Expression of the S to A mutant Lck-STIM1-C construct in HEK293 cells. (A) Representative live confocal image of cells expressing the mutant Lck-STIM1-CT construct bearing a C-terminal eGFP-tag. (B) Mean (± SE) AA-activated ARC channel currents measured at -80 mV in siRNA-treated cells expressing the siRNA-resistant Lck-STIM1-C construct (black), or the same siRNA-treated cells expressing the S to A mutant Lck-STIM1-C (gray). (C) The corresponding mean (± SE) I/V curves for the Lck-STIM1-C-expressing cells (black) and the mutant Lck-STIM1-C-expressing cells (gray). (D) Representative western blot showing relative expression of eGFP-tagged wild-type Lck-STIM1-C and the S to A mutant Lck-STIM1-C levels transfected into cells, as assessed by an eGFP Ab.
Reversing the orientation of the PM-associated STIM1 C terminal
We have shown that the cytosolic portion of STIM1, when anchored by its N terminal to the inner surface of the plasma membrane, fully supports the normal activation of the ARC channels, but fails to allow activation of the co-existing CRAC channels following store-depletion. One possibility for this failure could be that the normal interactions between CRAC channels and the STIM1 in the ER would involve the cytosolic portion of STIM1 being presented to the Orai in the plasma membrane in the “reverse” orientation, i.e., with the C terminus approaching the inner surface of the plasma membrane. In this context, studies have suggested that a polybasic region at the extreme C-terminal end of STIM1 interacts with membrane phosphoinositides to stabilize interactions of ER-resident STIM1 with the plasma membrane.Citation11,Citation13 Given this, we considered whether tethering the STIM1-CT construct to the plasma membrane in the reverse orientation using a C-terminal membrane-associating construct, might allow activation of the CRAC channels. To achieve this, we utilized the C-terminal domain of K-ras4B that has been shown to be critical in selective targeting of the protein to the plasma membrane.Citation34 This domain comprises a terminal CAAX box (C-V-I-M) preceded by a polybasic sequence containing eight lysine residues. During processing in the cell, the terminal V-I-M sequence of the CAAX box is deleted and the remaining cysteine becomes farnesylated. This prenylation, combined with the presence of the polybasic C-terminal residues, represents the critical sequence for targeting to the plasma membrane.Citation34,Citation35 Interestingly, as noted above, the C-terminal sequence of STIM1 contains a similarly lysine-rich polybasic sequence. We therefore deleted the first two lysines from the above K-ras4B CAAX box and attached the remaining sequence to the C-terminal end of the siRNA-resistant STIM1-CT construct to form a “STIM1-C-CAAX” construct.
As with the previous STIM1-CT constructs, expression of the siRNA-resistant STIM1-C-CAAX construct in STIM1 siRNA-treated cells failed to result in the appearance of any detectible constitutive currents. More importantly, use of the adenophostin-A/Ca2+-free pipette solution to maximally deplete the intracellular Ca2+ stores in these STIM1-C-CAAX-expressing cells failed to result in the development of any significant CRAC channel currents (inward current at -80 mV = 0.22 ± 0.03 pA/pF, n = 5). Western blot analysis of cells transfected with the STIM1-C-CAAX construct confirmed that this failure to enable the normal, store-dependent activation of the endogenous CRAC channels was not due to any lack of expression (Fig. S1A). Moreover, confocal imaging of cells expressing this construct bearing an N-terminal eGFP tag clearly revealed the predicted plasma membrane localization of the STIM1-C-CAAX protein (Fig. S1B). To confirm this apparent inability of the STIM1-C-CAAX construct to support the activation of endogenous CRAC channels, we again turned to the significantly larger CRAC channel currents in RBL-1 cells. However, as with the HEK cells, maximal depletion of internal Ca2+ stores by application of adenophostin-A/Ca2+-free pipette solution in RBL-1 cells expressing this construct resulted in inward currents measured at -80 mV of only 0.16 ± 0.03 pA/pF (n = 5). This represents a value equivalent to only ~10% of the corresponding currents recorded in untransfected RBL-1 cells (see above). Clearly, whatever the precise basis for this failure to activate the CRAC channels, simply reversing the orientation of the plasma membrane-tethered STIM1-CT to attempt to mimic the way in which the C terminus of STIM1 in the ER is normally presented to the CRAC channels, is not sufficient to enable this portion of the STIM1 molecule to activate the channels.
Interestingly, and in marked contrast to the data obtained with the Lck-STIM1-C construct, experiments in Flp-In HEK293 cells revealed that addition of exogenous arachidonic acid (8 µM) to the STIM1-C-CAAX expressing cells also failed to induce any significant activation of the ARC channels (inward current at -80 mV = 0.14 ± 0.02 pA/pF, n = 5). This marked difference in the ability of the STIM1-C-CAAX and Lck-STIM1-C constructs to enable arachidonic acid-dependent activation of the endogenous ARC channels is certainly intriguing, although its precise basis is unclear.
Agonist activation and Lck-STIM1-C
Up to now, we have demonstrated that the expression of the Lck-STIM1-C construct results in a unique, selective arachidonic acid-dependent activation of the ARC channels. Of course, activation of the channel by exogenous application of arachidonic acid cannot be considered as truly physiological, as the normal receptor-mediated pathways are bypassed. To address this concern, we examined whether the Lck-STIM1-C construct was able to support the same specific activation of ARC channels under agonist-stimulated conditions. To do this, we used an approach based on our previous finding that, in cells in which the cytosolic calcium is clamped at ~100 nM by appropriate buffering of the patch pipette solution, store-operated CRAC channel currents and arachidonic acid-regulated ARC channel currents are strictly additive when activated independently and sequentially in the same cell.Citation36 Based on this, we compared total agonist-activated calcium currents in a HEK cell line stably expressing an inducible m3 muscarinic receptor with the corresponding agonist-activated currents in the same cells in which CRAC channel currents had already been maximally activated by incorporation of adenophostin-A (2 µM) in the patch pipette solution.
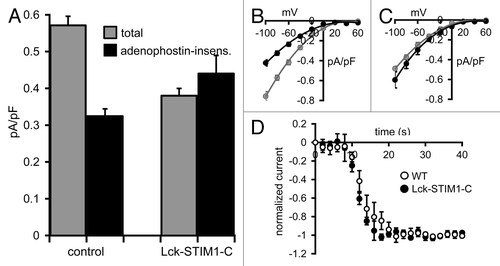
Initial Ca2+-imaging experiments using fura2-loaded cells revealed that addition of the muscarinic agonist carbachol (10 µM) resulted in a marked and prolonged increase in cytosolic Ca2+, consistent with an initial release of intracellular Ca2+ stores followed by a sustained Ca2+ entry (Fig. S2). In these m3-receptor expressing cells, whole-cell patch-clamp analysis showed that total agonist-activated currents averaged 0.57 ± 0.02 pA/pF (n = 6) at -80 mV in untransfected cells, a value that was decreased to 0.32 ± 0.02 pA/pF (n = 6) after prior maximal activation of the CRAC channels by exposure to internal adenophostin-A (). Assuming that the agonist-activated currents recorded after adenophostin-A treatment reflect those carried by the ARC channels, the data indicate that the ARC channel currents represent approximately 56% of the total agonist-activated currents in these cells. The identical procedure performed in STIM1 siRNA-treated cells expressing the siRNA-resistant Lck-STIM1-C construct resulted in total agonist-activated currents of 0.38 ± 0.02 pA/pF (n = 6) at -80 mV in the control cells, and 0.44 ± 0.05 pA/pF (n = 6) in the adenophostin-A pretreated cells (). These values were not statistically different from each other (p = 0.25), indicating that they specifically represented the magnitude of the ARC channel activity in each case. Critically, these data are entirely consistent with the proposal that the expression of the Lck-STIM1-C construct in siRNA-treated cells results in the ability of normal, agonist-mediated responses to induce the exclusive activation of the ARC channels. Finally, to examine whether the exclusive activation of the ARC channels by the Lck-STIM1-C construct influenced the rate of agonist-induced activation, we compared activation rates following addition of carbachol (10 µM) in the Lck-STIM1-C expressing m3-HEK cells with that in the wild-type m3-HEK cells. Normalizing the agonist-induced activation of current to the maximum current achieved in each case allowed direct comparison of the respective rates of activation. The data obtained () showed that there was no obvious difference in the rates of activation in each case.
Figure 5. Carbachol-activated currents in HEK cells expressing the Lck-STIM1-C construct. (A) Mean (± SE) carbachol-activated Ca2+ currents measured at -80 mV in HEK cells expressing the m3 muscarinic receptor. Shown are the total agonist-activated currents (gray), and the same currents recorded in cells after depletion of ER Ca2+ stores by intracellular adenophostin A (black) in untransfected control cells (control) and in STIM1 siRNA-treated cells expressing the Lck-STIM1-C construct. (B) Mean (± SE) I/V curves for the total agonist-activated currents (gray), and adenophostin-insensitive currents (black) in control cells and (C) the same for the Lck-STIM1-C expressing cells (see text for details). (D) Mean (± SE) curves comparing the activation kinetics of carbachol-activated currents in m3-HEK cells expressing the Lck-STIM1-C construct compared with those in wild-type m3-HEK cells. Total currents were normalized to their final maximal values to allow direct comparison of their respective rates of activation following addition of carbachol (10 µM) at time zero.
Discussion
Although the current emphasis on STIM1 as a regulator of store-operated CRAC channels focuses on its role in the ER, it was originally identified as a plasma membrane protein,Citation16,Citation37 and current evidence indicates that some 15–25% of the total pool of cellular STIM1 constitutively resides in the plasma membrane. Initial studies suggested that this pool might play a role in CRAC channel activation,Citation38 or that STIM1 was inserted into the plasma membrane as part of the activation of the CRAC channel following store-depletion.Citation7,Citation19 However, subsequent studies showed that any such insertion was not required for normal activation of the channels,Citation9,Citation15,Citation39 while other studies have challenged whether such insertion occurs at all.Citation6,Citation8,Citation10,Citation40 In contrast to the above, the essential role of plasma membrane STIM1 in the activation of the store-independent ARC channels seems clear,Citation15 although exactly how such activation occurs remains unknown.
With regard to the CRAC channels, current understanding of the means by which STIM1 in the ER activates these channels indicates that the N-terminal regions of the protein located in the lumen of the ER act to sense the depletion of Ca2+ from these intracellular Ca2+ stores, and then initiate the oligomerization of STIM1 prior to its translocation within the ER membrane to form puncta at sites close to the plasma membrane where it can interact with, and activate, the CRAC channels.Citation6,Citation8,Citation9,Citation11 Thus, although actual activation of the channels relies on the cytosolic regions of STIM1,Citation20,Citation21 it is the luminal N terminal part of the protein that is critical to the physiological initiation of this activation process. This region contains two distinct highly conserved domains, a Ca2+-binding EF-hand domain and an adjacent sterile α motif (SAM) domain.Citation17,Citation41 Curiously, it has been demonstrated that the former actually consists of two EF-hands, a canonical EF-hand that binds Ca2+ and a “hidden” EF-hand that does not bind Ca2+, but which has an important role in stabilizing interactions between the EF-hand and the SAM domain.Citation41 Studies using an isolated recombinant protein fragment consisting of the EF-hand and SAM domains (EF-SAM) showed that, with Ca2+ bound to the EF-hand, the EF-SAM peptide forms as a relatively stable, well-folded globular monomer. Loss of Ca2+ from the EF-hand, such as would occur on depletion of the ER stores, results in the destabilization and partial unfolding of this structure, which allows the subsequent dimerization and oligomerization of this region.Citation17,Citation41 Additional studies have demonstrated that this dimerization is, in itself, entirely sufficient to induce the complete sequence of subsequent events, including STIM1 oligomerization and translocation to sites close to the plasma membrane, which result in the activation of the channels.Citation12,Citation21 As such, with Ca2+ bound to the EF-hand, the intramolecular interactions between this domain and the SAM domain essentially act to inhibit the inherent intermolecular interactions between adjacent STIM1 molecules that are necessary for activation of the channels.Citation18 Critically, for STIM1 residing in the plasma membrane, the EF-hand is continually exposed to relatively stable, extracellular Ca2+ concentrations of ~2 mM, and, based on the above, would be expected to constitutively remain in this inhibitory state. Indeed, such an effect is entirely consistent with the demonstrated lack of any role for the endogenous plasma membrane-located STIM1 in the activation of the store-operated CRAC channels, as discussed above. In marked contrast however, the data presented here indicate that N-terminal regions of STIM1 (including the EF-hand and SAM domains) play no significant role in the arachidonic acid-dependent activation of the endogenous ARC channels. Thus, although the free cytosolic STIM1 C terminus is entirely inactive in itself, simply anchoring this construct to the inner face of the plasma membrane renders it fully capable of activating, in an exclusive manner, the store-independent ARC channels. However, this activation is not constitutive as it still requires arachidonic acid, either applied exogenously, or generated physiologically as a result of agonist action at a relevant receptor.
In conclusion, the data obtained indicate that, with STIM1 located in the plasma membrane, increases in arachidonic acid are able to overcome the inherent inhibitory effect of the Ca2+-bound EF-hand to allow activation of the endogenous channels. As yet, we do not know how this might be achieved, and several possibilities exist. For example, arachidonic acid may directly induce interactions between the channels and STIM1 in the plasma membrane, circumventing the normal inhibitory effect of the Ca2+-bound N-terminal domains. Alternatively, perhaps as a result of the localization of both the STIM1 and the channel in the same membrane and the consequent “higher effective concentration” in this two-dimensional surface,Citation21 the two may be constitutively associated with each other, and arachidonic acid acts on this preformed complex to activate the channel. In any event, although it is clear that the STIM1 C terminus is responsible for the activation of both CRAC and ARC channels, it remains to be determined whether the specific inter- and intramolecular interactions that underlie the activation of the Orai proteins that form the respective channels are similar in these two distinct conditions. The development of the unique STIM1 construct described here, which enables the separate and exclusive activation of the ARC channels under arachidonic acid-mediated conditions as well as following physiologically relevant agonist-activated action, provides an important tool in identifying such differences. Moreover, the co-existing CRAC and ARC channels appear to play distinct roles in the regulation of certain calcium-dependent cellular activities. As an example, it has recently been shown that the ARC channels uniquely modulate the frequency of agonist-activated calcium oscillations by inducing the local activation of a member of the phospholipase C delta family.Citation42 Consequently, this construct should prove useful in delineating the specific physiological and/or pathophysiological roles of these channels in the various cell types in which they are found.
Materials and Methods
Cells and constructs
Flp-In HEK293 cells (Invitrogen) and RBL-1 cells (ATCC) were maintained in EMEM (ATCC) plus 10% FBS in an incubator at 37°C, gassed with 5% CO2 in air. Depletion of endogenous STIM1 in the Flp-In cells was attained using siRNA duplexes (225 pmol/100 μl) targeting the AGGTGGAGGTGCAATATTA sequence in human STIM1 (Dharmacon Inc.) as previously described.Citation15 For the RBL-1 cells, siRNA duplexes were made targeting the same region within the rat STIM1 sequence. An siRNA-resistant STIM1 was generated by making silent mutations to two bases (G–A and G–C) in the siRNA recognition site of a human STIM1 pcmv6-XL5 construct (OriGene) using QuikChange II (Stratagene). This siRNA-resistant STIM1 construct was then used as the template for all the remaining constructs used in this study (see Supplementary Material). All constructs were nucleofected using an Nucleofector (Amaxa) using either 0.5 µg (Flp-In) or 2 µg (RBL-1) of plasmid DNA and/or 225 pmol of STIM1 siRNA 48 h prior to use. The HEK293 cell line stably expressing an inducible m3 muscarinic receptor was generated using the Flp-ln T-Rex system (Invitrogen) and maintained as above. Cells were induced for 24 h prior to experimentation with 1 µg/ml tetracycline.
Western blot analysis
Transfected cells were washed with Ca2+/Mg2+-free PBS, lysed in 25 mM TRIS-HCl pH 7.6, 150 mM NaCl, 1% sodium deoxycholate and 0.1% SDS, plus a mini complete protease inhibitor tablet (Roche), sonicated and spun at 14,000 g at 4°C for 15 min. Supernatants were run on a 7% SDS-PAGE gel, transferred onto nitrocellulose and probed with a STIM1 C-terminal pAb (Cell Signaling) followed by IRDye 800CW anti-rabbit secondary Ab, or eGFP mAb (Clontech) followed by IRDye 800CW anti-mouse secondary Ab. Blots were stripped and reprobed with a β-actin mAb (Sigma) followed by IRDye 800CW anti mouse-secondary Ab. All blots were analyzed on an Odyssey Scanner (Li-Cor).
Electrophysiology
For patch-clamp experiments, cells were plated onto 5 mm polylysine-coated coverslips for 18–24 h prior to study. Whole-cell current recordings were performed at 20–22°C, using 250 ms voltage pulses to -80 mV delivered every 2 sec from a holding potential of 0 mV. Current-voltage relationships were obtained by applying 10 ms pulses to potentials between -100 mV and +60 mV at 20 mV intervals. All patch-clamp experiments were performed with expression of constructs without an attached eGFP tag. This tag was only added for the purposes of confocal analysis of the subcellular distribution of the expressed constructs (see below). To permit identification of transfected cells under appropriate illumination, cells were co-transfected with a pVenus reporter construct. Transfection efficiency was typically ~65–70% for the Flp-In HEK cells, but significantly less (~25–30%) for the RBL cells. Arachidonic acid-activated currents were determined after switching to a bath solution containing arachidonic acid (8 μM). Initial currents obtained before activation of the channel were used for leak subtraction of subsequent recordings. Store-operated currents were recorded using a Ca2+-free internal (pipette) solution, which contained the potent InsP3 receptor agonist adenophostin A (2 μM). Because of potential uncertainties as to when these currents were initiated on achieving the whole-cell configuration, currents obtained at the end of each experiment using an external solution containing La3+ (100 μM) were used for leak-subtraction. See Supplementary Material for the composition of the extracellular and intracellular (pipette) solutions.
Confocal imaging
For confocal microscopy, the relevant constructs were terminally tagged with an attached eGFP. Cells were plated onto 25 mm coverslips 18 h prior to imaging on a C1 laser scanning confocal microscope (Nikon) with a 40x (1.3 NA) Fluor oil immersion objective. To image eGFP, excitation at 488-nm was obtained using an argon laser line, with emission collected via a 543-nm bandpass filter and photomultiplier tube.
Single-cell Ca2+ measurements
Agonist-induced changes in cytosolic Ca2+ ion concentrations were performed on Flp-In HEK293 cells stably expressing the muscarinic m3 receptor following intracellular loading with fura2, as previously described.Citation42
| Abbreviations: | ||
| ARC channel | = | arachidonate-regulated Ca2+ channel |
| CRAC channel | = | Ca2+ release-activated Ca2+ channel |
| STIM1 | = | stromal interacting molecule 1 |
Additional material
Download Zip (199.2 KB)Acknowledgments
We thank Drs David Yule, Alan Smrcka and Greg Tall of the Department of Pharmacology and Physiology, University of Rochester Medical Center for helpful discussions and advice and P. Leakey for technical assistance. This work was supported by National Institutes of Health Grant GM040457 to T.J.S.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/channels/article/21947/
References
- Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, et al. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA 2008; 105:13668 - 73; http://dx.doi.org/10.1073/pnas.0806499105; PMID: 18757751
- Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol 2008; 586:419 - 25; http://dx.doi.org/10.1113/jphysiol.2007.147249; PMID: 18006576
- Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature 2008; 456:116 - 20; http://dx.doi.org/10.1038/nature07338; PMID: 18820677
- Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol 2008; 586:185 - 95; http://dx.doi.org/10.1113/jphysiol.2007.146258; PMID: 17991693
- Mignen O, Thompson JL, Shuttleworth TJ. The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of Orai1 and Orai3 subunits. J Physiol 2009; 587:4181 - 97; http://dx.doi.org/10.1113/jphysiol.2009.174193; PMID: 19622606
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 2005; 15:1235 - 41; http://dx.doi.org/10.1016/j.cub.2005.05.055; PMID: 16005298
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005; 437:902 - 5; http://dx.doi.org/10.1038/nature04147; PMID: 16208375
- Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 2006; 174:803 - 13; http://dx.doi.org/10.1083/jcb.200604014; PMID: 16966422
- Baba Y, Hayashi K, Fujii Y, Mizushima A, Watarai H, Wakamori M, et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA 2006; 103:16704 - 9; http://dx.doi.org/10.1073/pnas.0608358103; PMID: 17075073
- Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, et al. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem 2006; 281:24979 - 90; http://dx.doi.org/10.1074/jbc.M604589200; PMID: 16807233
- Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 2007; 104:9301 - 6; http://dx.doi.org/10.1073/pnas.0702866104; PMID: 17517596
- Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008; 454:538 - 42; http://dx.doi.org/10.1038/nature07065; PMID: 18596693
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009; 136:876 - 90; http://dx.doi.org/10.1016/j.cell.2009.02.014; PMID: 19249086
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 2009; 11:337 - 43; http://dx.doi.org/10.1038/ncb1842; PMID: 19182790
- Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol 2007; 579:703 - 15; http://dx.doi.org/10.1113/jphysiol.2006.122432; PMID: 17158173
- Manji SS, Parker NJ, Williams RT, van Stekelenburg L, Pearson RB, Dziadek M, et al. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta 2000; 1481:147 - 55; http://dx.doi.org/10.1016/S0167-4838(00)00105-9; PMID: 11004585
- Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem 2006; 281:35855 - 62; http://dx.doi.org/10.1074/jbc.M608247200; PMID: 17020874
- Zheng L, Stathopulos PB, Schindl R, Li GY, Romanin C, Ikura M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc Natl Acad Sci USA 2011; 108:1337 - 42; http://dx.doi.org/10.1073/pnas.1015125108; PMID: 21217057
- Hauser CT, Tsien RY. A hexahistidine-Zn2+-dye label reveals STIM1 surface exposure. Proc Natl Acad Sci USA 2007; 104:3693 - 7; http://dx.doi.org/10.1073/pnas.0611713104; PMID: 17360414
- Muik M, Fahrner M, Derler I, Schindl R, Bergsmann J, Frischauf I, et al. A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J Biol Chem 2009; 284:8421 - 6; http://dx.doi.org/10.1074/jbc.C800229200; PMID: 19189966
- Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell 2010; 21:1897 - 907; http://dx.doi.org/10.1091/mbc.E10-02-0145; PMID: 20375143
- Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, et al. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat Cell Biol 2006; 8:1003 - 10; http://dx.doi.org/10.1038/ncb1454; PMID: 16906149
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem 2008; 283:8014 - 22; http://dx.doi.org/10.1074/jbc.M708898200; PMID: 18187424
- Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, et al. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem 2008; 283:17662 - 71; http://dx.doi.org/10.1074/jbc.M801536200; PMID: 18420579
- Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun 2009; 385:49 - 54; http://dx.doi.org/10.1016/j.bbrc.2009.05.020; PMID: 19433061
- Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem 2007; 282:29448 - 56; http://dx.doi.org/10.1074/jbc.M703573200; PMID: 17702753
- Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, Ritchie MF, et al. STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci USA 2009; 106:7391 - 6; http://dx.doi.org/10.1073/pnas.0900293106; PMID: 19376967
- Koegl M, Zlatkine P, Ley SC, Courtneidge SA, Magee AI. Palmitoylation of multiple Src-family kinases at a homologous N-terminal motif. Biochem J 1994; 303:749 - 53; PMID: 7980442
- Kwong J, Lublin DM. Amino-terminal palmitate or polybasic domain can provide required second signal to myristate for membrane binding of p56lck. Biochem Biophys Res Commun 1995; 207:868 - 76; http://dx.doi.org/10.1006/bbrc.1995.1266; PMID: 7864883
- Bijlmakers MJ, Isobe-Nakamura M, Ruddock LJ, Marsh M. Intrinsic signals in the unique domain target p56(lck) to the plasma membrane independently of CD4. J Cell Biol 1997; 137:1029 - 40; http://dx.doi.org/10.1083/jcb.137.5.1029; PMID: 9166404
- Zlatkine P, Mehul B, Magee AI. Retargeting of cytosolic proteins to the plasma membrane by the Lck protein tyrosine kinase dual acylation motif. J Cell Sci 1997; 110:673 - 9; PMID: 9092949
- McCabe JB, Berthiaume LG. Functional roles for fatty acylated amino-terminal domains in subcellular localization. Mol Biol Cell 1999; 10:3771 - 86; PMID: 10564270
- Yasuda K, Kosugi A, Hayashi F, Saitoh S, Nagafuku M, Mori Y, et al. Serine 6 of Lck tyrosine kinase: a critical site for Lck myristoylation, membrane localization, and function in T lymphocytes. J Immunol 2000; 165:3226 - 31; PMID: 10975838
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990; 63:133 - 9; http://dx.doi.org/10.1016/0092-8674(90)90294-O; PMID: 2208277
- Roy MO, Leventis R, Silvius JR. Mutational and biochemical analysis of plasma membrane targeting mediated by the farnesylated, polybasic carboxy terminus of K-ras4B. Biochemistry 2000; 39:8298 - 307; http://dx.doi.org/10.1021/bi000512q; PMID: 10889039
- Mignen O, Shuttleworth TJ. I(ARC), a novel arachidonate-regulated, noncapacitative Ca(2+) entry channel. J Biol Chem 2000; 275:9114 - 9; http://dx.doi.org/10.1074/jbc.275.13.9114; PMID: 10734044
- Williams RT, Senior PV, Van Stekelenburg L, Layton JE, Smith PJ, Dziadek MA. Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta 2002; 1596:131 - 7; http://dx.doi.org/10.1016/S0167-4838(02)00211-X; PMID: 11983428
- Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci USA 2006; 103:4040 - 5; http://dx.doi.org/10.1073/pnas.0510050103; PMID: 16537481
- Hewavitharana T, Deng X, Wang Y, Ritchie MF, Girish GV, Soboloff J, et al. Location and function of STIM1 in the activation of Ca2+ entry signals. J Biol Chem 2008; 283:26252 - 62; http://dx.doi.org/10.1074/jbc.M802239200; PMID: 18635545
- Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol 2006; 16:1465 - 70; http://dx.doi.org/10.1016/j.cub.2006.05.051; PMID: 16860747
- Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008; 135:110 - 22; http://dx.doi.org/10.1016/j.cell.2008.08.006; PMID: 18854159
- Thompson JL, Shuttleworth TJ. Orai channel-dependent activation of phospholipase C-δ: a novel mechanism for the effects of calcium entry on calcium oscillations. J Physiol 2011; 589:5057 - 69; http://dx.doi.org/10.1113/jphysiol.2011.214437; PMID: 21878525