Abstract
Mangroves are complex and dynamic ecosystems varying in salinity, water level and nutrient availability; they also contain diverse and distinct microbial communities. Studies of microbes and their interactions with other ecosystem components (e.g., tree roots) are critical for our understanding of mangrove ecosystem functioning and remediation. Using a barcoding pyrosequencing approach, we previously noted the persistence of terrestrial bacterial populations on mangrove roots when nursery raised saplings were transplanted back to their natural environment. Here we go into further detail about the potential functional associations of bacterial guilds with distinct mangrove microhabitats including the rhizosphere. We also use a nonparametric richness estimator to show that estimated operational taxonomic unit (OTU) richness is more than twice that observed. In the transplant microhabitat, our estimate suggests that there are almost 7000 OTU's for a sample size of 10400 individual sequences with no sign of an asymptote, indicating that 'true' richness for this microhabitat is substantially larger. Results on the number of bacterial OTU's should, however, be viewed with caution given that the barcoding pyrosequencing technique used can yield sequencing artifacts that may inflate richness estimates if not properly removed.
Mangroves are among the most productive ecosystems in the world and are of ecological, economic and societal importance.Citation1 Microbes play a key role in maintaining this productivity;Citation2 in fact they also constitute the largest pool of metabolic pathways on Earth with potential biotechnological and environmental implications. However, the benefits of microorganisms (in particular those associated with roots) to the productivity of mangroves and the roles they play in plant fitness, survival and overall ecosystem resilience have been scarcely studied. Unveiling the diversity and structure of microbial communities in mangrove environments represents the first step towards a better understanding of their role in ecosystem functioning.
Coupling Restoration Strategies with Molecular Microbial Ecology
Multidisciplinary and integrated research approaches can provide the means for understanding and managing highly complex coastal ecosystems such as mangrove forests. In a recent study,Citation3 we presented data on bacterial composition in a mangrove ecosystem located nearby Rio de Janeiro, Brazil. A reforestation plan was implemented, which aimed at transplanting Rhizophora mangle saplings from terrestrial nursery settings to a perturbed mangrove area. We monitored the persistence of bacterial populations in the rhizospheres—i.e., microhabitats influenced by plant roots—of healthy individuals 202 days after transplantation, and compared bacterial composition in bulk sediment and the rhizospheres of transplanted and native R. mangle saplings. A massively parallel sequencing technology (i.e., 454 pyrosequencing) was used to assess the composition of bacterial 16S rRNA gene amplicons retrieved from rhizosphere and sediment metagenomic DNA. This method circumvents the drawbacks of previous molecular techniques and importantly enables sequencing of tens to hundreds of millions of base pairs in a typical run.Citation4 When combined with experimental manipulations in situ or controlled microcosms, the true advantages of using next-generation sequencing in environmental microbiology will come into play.
Associations between Bacterial Taxa and Mangrove Microhabitats
In our previous study,Citation3 we drew attention to apparent associations between various bacterial taxa and the mangrove rhizosphere. Among other things we noticed that Rhizobiales, Campylobacterales, Methylococcales and Vibrionales tended to be more abundant in the rhizosphere samples than in the bulk sediment.Citation3 One association of particular interest concerned Vibrio species. Vibrio spp. are known for their well documented associations with metazoans, their occurrence as free-living commensals in the marine realm and the prevalence of some pathogenic taxa. The recent cultivation of nitrogen-fixing Vibrio spp. from salt marsh and mangrove rhizospheres suggests that these species might be involved in a hitherto unknown but potentially important interaction with plants.Citation5,Citation6 In our study,Citation3 we observed a clear augmentation of 16S rRNA gene tags assigned to Vibrio spp. in native rhizosphere vs. bulk sediment samples, as well as in transplanted vs. nursery rhizosphere samples. Interestingly, one OTU assigned to Vibrio spp. was the most abundant detected in the mangrove rhizosphere samples (363 reads).Citation3 This has important implications for our understanding of the niche breadth of Vibrio species in nature. Comparative genomics will possibly enlighten our perspective of the plasticity of Vibrio genomes and gene clusters thereof that provide them with a fitness-enhancing factor in the rhizosphere environment, as well as sharpen our understanding of common and specific traits involved in rhizosphere competence in terrestrial vs. marine biomes.
Estimating Bacterial Richness in Mangrove Sediments
Pyrosequencing produces orders of magnitude more data than previous techniques but still only covers a fraction of the total bacterial community.Citation7 Because of this, various studies have attempted to estimate “true” richness.Citation8,Citation9 There are various ways of achieving this goal, including extrapolating accumulation curves,Citation10 parametric model fittingCitation8 or using nonparametric richness estimators.Citation11,Citation12 Nonparametric richness estimators have the advantage that no assumptions need to be made about community structure, as opposed to parametric models. Comparative studies have also shown that some nonparametric estimators outperform other non-parametric and parametric estimators. Colwell and Coddington,Citation11 for example, found that the Chao1 and Chao2 estimators provided the least biased estimates of species richness for small numbers of samples. Likewise, Bent and ForneyCitation12 found that nonparametric estimators including Chao1 produced the most accurate estimate of microbial diversity from clone library data. The ACE, Chao1, Jackknife1 and Bootstrap also performed well in a comparison with 12 richness estimators used to estimate the species richness of epigean arthropods in Azorean forests.Citation13 It is important to note, however, that estimations produced using nonparametric richness estimators represent “minimum” estimations. The values should thus be seen as lower bounds of total species richness given the data in the sample set.Citation14
In addition to the above, barcoding pyrosequencing typically delivers sequencing artifacts that might inflate bacterial richness if not properly removed from the dataset. Stringent quality-based sequence trimming and adequate choice of clustering cut-off levels are essential for more reliable richness estimations.Citation15,Citation16
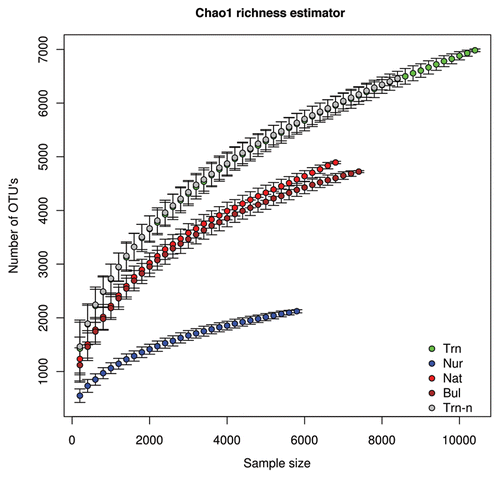
Here we use the Chao1 species richness estimatorCitation17 to estimate the total number of bacterial OTU's in rhizosphere and bulk sediment samples. These include soil from four “microhabitats,” namely (1) the roots of nursery plants before planting in a mangrove (Nur), (2) roots of transplanted saplings 202 dap in a mangrove (Trn), (3) roots of native (non-transplanted) saplings in a mangrove (Nat) and (4) bulk sediment in the mangrove replant area (see Gomes et al.Citation3 for detailed description of the samples). (observed OTU richness) and (estimated OTU richness) show the number of bacterial OTU's—defined by 97% sequence similarity between 16S rRNA gene fragments spanning the V4 region of the gene—estimated as a function of sample size using the rarefaction and the Chao1 richness estimator.
The maximum total number of OTU's was found in the transplant microhabitat, namely 3659.39 ± 2.62 OTU's for 10,400 individual sequences (). Controlling for sample size (n = 5,800 individual sequences), OTU richness varied from 1243.50 ± 3.65 for the nursery microhabitat, to 2244.10 ± 14.50 for the bulk microhabitat, 2284.04 ± 12.15 for the native microhabitat and finally 2600.56 ± 18.33 for the transplant microhabitat.
Qualitatively, the Chao1 estimator reveals a similar pattern to observed OTU richness, namely a substantially reduced pool of OTU's in nursery samples and the largest number of OTU's in transplants in line with results presented in our previous study in reference Citation3. The number of OTU's is only slightly higher in the native microhabitat than the bulk soil microhabitat with both the observed and estimated OTU richness. Estimated OTU richness using the Chao1 estimator is still, however, substantially higher than what was observed. The transplant microhabitat had the highest estimated OTU richness, namely, (6984.55 ± 25.38 for 10400 individual sequences), more than twice the number observed (). None of the microhabitats, however, showed signs of reaching an asymptote over a sample size of 5,800 to 10,000 individual sequences, indicating that “true” OTU richness is even higher.
Samples from the nursery and transplants shared a total of 364 OTU's (thus representing almost 10% of the OTU's found in the transplants); this included a number of abundant OTU's described in Gomes et al.Citation3 Initially, we expected that the markedly higher OTU richness found in transplants was due to species from the nursery augmenting the richness of transplants. In order to test this hypothesis, we removed all OTU's shared between the nursery and the transplants, and recalculated observed and expected OTU richness for the transplants (Trn-n in and ). Contrary to expectations, this had very little effect on observed or estimated OTU richness after controlling for sample size ( and ). This somewhat surprising result indicates that an augmentation by nursery microbes does not appear to be the main cause of the high OTU richness in transplants, although it does appear to affect overall microbe abundance and indeed composition as shown in Gomes et al.Citation3 This begs the question of what exactly is responsible for this augmentation. Unfortunately, there are no clear cut answers. It should be noted that most of the OTU's found in the transplants were in fact only found there (1,915 out of 3,668: 52% of OTU's). The percentage of OTU's restricted to the nursery was higher (868 out of 1,258: 69%), but overall richness much lower. In the mangrove environment itself 45% (1,120 out of 2,513) and 47% (1,204 out of 2,585) of OTU's were only found in the native and bulk soil microhabitats, respectively. Although speculative, it is possible that the conditions to which the transplants were subjected (e.g., initial growth in the nursery combined with subsequent transplantation into the very different mangrove environment, transportation, residual nursery soil attached to the roots, root density due to artificial growth conditions and replanting) created a uniquely heterogeneous root environment allowing the establishment of a particularly rich and densely packed microbial community.
Concluding Remarks and Outlook
In the last few years, 16S rRNA gene pyro-sequencing has become a common tool to study bacterial diversity. Interestingly, richness values inferred here compare well with those reported by Hollister et al.Citation18 who used a similar sequencing effort in the analysis of a range of soil and sediment samples of a hypersaline lake located in Southern Texas. For example, the Chao 1 richness estimates for the water-logged sediment samples,Citation18 were similar to values (4,556 up to 5,285) found in this study for bulk sediment samples (). It is curious to note that such estimates fit relatively well with those proposed for soils and sediments on the basis of “sequence-less,” DNA reassociation data.Citation19,Citation20 Richness estimates predicted in these seminal surveys have become an influential “10,000-bacterial-genomes-in-one-gram-of-soil” paradigm that permeated mainstream thinking in last decade's microbial ecology. Recent improvements in the analysis of DNA reassociation rates have, however, suggested that up to 1 million different genomes could theoretically coexist in a typical sample.Citation21 The latter view has been a matter of great debate. To date, hard data derived from high-throughput 16S rRNA gene sequencing seems closer to the 104 rather than the 106 view of bacterial species richness per sample. It, however, remains to be understood how well 16S rRNA gene data translate into whole-genome bacterial diversity and, likewise, the extent to which the latter translates into different bacterial species.
Figures and Tables
Figure 1 Bacterial richness in mangrove sediments and rhizospheres estimated using rarefaction. Four microhabitats were sampled: Nur, roots of nursery plants before planting in a mangrove; Trn, roots of transplanted saplings 202 days after planting in a mangrove; Trn-n, roots of transplanted saplings 202 days after planting without OTU's shared between the nursery and the transplants; Nat, roots of native (non-transplanted) saplings in a mangrove (Nat); and Bul, bulk sediment in the mangrove replant area. Four replicates per habitat were surveyed. The species accumulation curves shown represent values for pooled replicates.
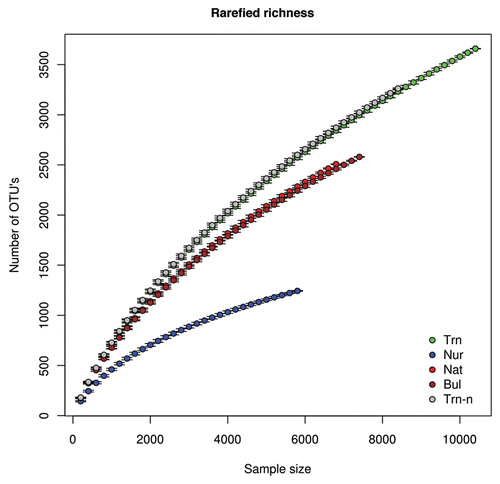
Figure 2 Bacterial richness in mangrove sediments and rhizospheres estimated with the Chao1 richness estimator. Four microhabitats were sampled: Nur, roots of nursery plants before planting in a mangrove; Trn, roots of transplanted saplings 202 days after planting in a mangrove; Trn-n, roots of transplanted saplings 202 days after planting without OTU's shared between the nursery and the transplants. Nat, roots of native (non-transplanted) saplings in a mangrove (Nat); and Bul, bulk sediment in the mangrove replant area. Four replicates per habitat were surveyed.
Acknowledgments
This study was funded by the German Research Foundation SM59/4-1 and 4-2 (www.dfg.de/en/index.jsp), FAPERJ-Brazil (www.faperj.br), Centre for Environmental and Marine Studies (CESAM, Portugal) (www.cesam.ua.pt) and Foundation for Science and Technology (FCT, Portugal) PTDC/AAC-CLI/107916/2008 (alfa.fct.mctes.pt).
Addendum to:
References
- Alongi DM. Present state and future of the world's mangrove forests. Environ Conserv 2002; 29:331 - 349
- Holguin G, Vazquez P, Bashan Y. The role of sediment microorganisms in the productivity, conservation and rehabilitation of mangrove ecosystems: an overview. Biol Fertil Soils 2001; 33:265 - 278
- Gomes NCM, Cleary DFR, Pinto FN, Egas C, Almeida A, Cunha A, et al. Taking root: Enduring effect of rhizosphere bacterial colonization in mangroves. PLoS ONE 2010; 5:14065; http://dx.doi.org/10.1371/journal.pone.0014065
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005; 437:376 - 380
- Criminger JD, Hazen TH, Sobecky PA, Lovell CR. Nitrogen fixation by Vibro parahaemolyticus and its implications for a new ecological niche. Appl Environ Microbiol 2007; 73:5959 - 5961
- Rameshkumar N, Nair S. Isolation and molecular characterization of genetically diverse antagonistic, diazotrophic red-pigmented vibrios from different mangrove rhizospheres. FEMS Microbiol Ecol 2009; 67:455 - 467
- Roesch L, Fulthorpe R, Riva A, Casella G, Hadwin A, Kent A, et al. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 2007; 1:283 - 290
- Curtis TP, Head IM, Lunn M, Woodcock S, Schloss PD, Sloan WT. What is the extent of prokaryotic diversity?. Philos Trans R Soc Lond B Biol Sci 2006; 361:2023 - 2037
- Bent SJ, Forney LJ. The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J 2008; 2:689 - 695
- Van Loon EE, Cleary DFR, Fauvelot C. ARES: software to compare allelic richness between uneven samples. Mol Ecol Notes 2007; 7:579 - 582
- Colwell RK, Coddington JA. Estimating terrestrial biodiversity through extrapolation. Philos Trans R Soc Lond B Biol Sci 1994; 345:101 - 118
- Bent SJ, Forney LJ. The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J 2008; 2:689 - 695
- Hortal J, Borges PAV, Gaspar C. Evaluating the performance of species richness estimators: sensitivity to sample grain size. J Anim Ecol 2006; 75:274 - 287
- Colwell RK. ESTIMATES: statistical estimation of species richness and shared species from samples. Version 75 2007;
- Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 2010; 12:118 - 123
- Schloss P. The Effects of Alignment Quality, Distance Calculation Method, Sequence Filtering and Region on the Analysis of 16S rRNA Gene-Based Studies. PLoS Comput Biol 2010; 6:1000844; http://dx.doi.org/10.1371/journal.pcbi.1000844
- Chao A, Chazdon RL, Colwell RK, Shen TJ. A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol Lett 2005; 8:148 - 159
- Hollister E, Engledow A, Hammett A, Provin T, Wilkinson H, Gentry T. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 2010; 4:829 - 838
- Torsvik V, Daae FL, Sandaa RA, Ovreas L. Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol 1998; 64:53 - 62
- Torsvik V, Goksoyr J, Daae FL. High diversity in DNA of soil bacteria. Appl Env Microbiol 1990; 56:782 - 787
- Gans J, Wolinsky M, Dunbar J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005; 309:1387 - 1390