Abstract
Nitric oxide (NO) is an endogenous second messenger which acts as a potent vasodilator, anti-inflammatory, anti-thrombotic, and pro-angiogenic agent in the vasculature. Recent studies revealed that the effects of NO on blood vessels are mediated in part by its ability to regulate protein trafficking machinery and vesicle-based exocytosis in vascular cells. Specifically, NO-dependent S-nitrosylation of N-ethylmaleimide sensitive factor (NSF), an ATPase that enables membrane fusion, was shown to inhibit exocytosis of vesicular secretory compartments such as endothelial Weibel-Palade bodies, platelet alpha granules, and cytolytic granules from activated lymphocytes. Tissue transglutaminase (tTG or TG2) is a multifunctional protein synthesized and secreted by various cell types in the vasculature, which is involved in multiple vascular diseases, including atherosclerosis, vascular calcification, and age-dependent aortic stiffening. Our recent findings indicate that tTG is delivered to the cell surface and the extracellular matrix (ECM) via a non-classical ER/Golgi-independent secretion pathway, which depends on the recycling endosomes and the NSF function. Here we report that NO attenuates the unconventional secretion of tTG in human aortic endothelial cells. NO-dependent down-regulation of extracellular tTG levels via inhibition of its secretion might be a part of general physiological mechanism which limits externalization of adhesive, pro-inflammatory and thrombogenic proteins in the vasculature.
tTG is a ubiquitously expressed member of transglutaminase family which displays the transamidating/protein cross-linking function, but, unlike other transglutaminases, also possesses GTPase and disulfide isomerase activities, as well as additional nonenzymatic adhesive/signaling functions.Citation1,Citation2 Among the latter, tTG is known to noncovalently bind and regulate signaling by integrins, syndecan-4 and platelet derived growth factor receptor (PDGFR).Citation3–Citation5 The majority of tTG is localized in the cytoplasm, yet it is externalized from undamaged endothelial and smooth muscle cells, monocytes/macrophages, fibroblasts and osteoblasts, which all contain this protein on their surface and in the surrounding ECM.Citation1 Since tTG does not have leader peptide or hydrophobic domains typical for secreted proteins,Citation6 this dual topology appears surprising. While tTG is associated with cellular membranes, it is not localized to the ER/Golgi compartments involved in classical protein secretion.Citation1,Citation2 Our latest studies showed that tTG is secreted via a constitutive non-classical pathway which includes phospholipid-mediated docking of cytoplasmic protein to perinuclear recycling endosomes, its delivery inside these transport vesicles, their anterograde movement and fusion with plasma membrane which releases intravesicular tTG onto the cell surface.Citation7 The functions of membrane fusion ATPase NSF, soluble NSF attachment receptor proteins (SNAREs) VAMP3 and SNAP23, and regulatory GTPase Rab11, appear all essential for tTG externalization. Citation7 While this secretion pathway is likely common for many cell types that express tTG, physiological mechanisms of its regulation remain unknown.
Studies by several laboratories began to unravel a complex role of tTG in vascular pathophysiology. The protein was found to contribute to pressure-induced inward remodeling in small arteries and both its enzymatic (protein cross-linking) and non-enzymatic functions are involved in this process.Citation8 tTG was also implicated in osteochondrogenic transdifferentiation of vascular smooth muscle cells in vitro and atherosclerotic calcification in human carotid arteries.Citation9,Citation10 In addition, the cross-linking tTG activity was shown to promote macrophage infiltration into atherosclerotic lesions.Citation11 Finally, this activity of tTG was reported to contribute to age-dependent increases in vascular stiffness in rats, and is likely to decrease arterial compliance in aging humans.Citation12
Nitric oxide is a key signaling molecule in the vasculature where it affects multiple aspects of blood vessel homeostasis, including vascular tone, leukocyte trafficking and platelet adhesion/aggregation.Citation13 Recent studies showed the ability of NO to regulate protein trafficking and secretion in vascular cells.Citation13,Citation14 NO-mediated S-nitrosylation of Cys264 in NSF does not affect the ATPase activity of this molecular motor, but blocks its disassembly from complexes with SNAREs and inhibits membrane fusion.Citation14 This regulation occurs in endothelial cells where NO inhibits secretion of Weibel-Palade bodies,Citation15 in platelets where it prevents externalization of dense, lysosomal and alpha granules,Citation16 and in activated T lymphocytes where it impedes exocytosis of cytolytic granules.Citation17 In addition, NO S-nitrosylates dynamin, increasing its oligomerization and GTPase activity, and elevating the rate of receptor-mediated endocytosis.Citation18 These NO-mediated modifications of the key regulators of protein trafficking alter cell surface proteome by modulating extracellular levels and activities of adhesive, pro-inflammatory and thrombogenic proteins in vascular cells.Citation13
Is tTG a target of NO-mediated regulation in the vasculature? Greenberg and colleagues showed that the cross-linking activity of tTG is inhibited by S-nitrosylation in vitroCitation19 and we found that endogenously produced NO suppresses tTG-mediated transamidation/cross-linking in human aortic endothelial cells (HAECs) and in mouse aorta.Citation12 In agreement with diminished bioavailability of NO in aging blood vessels, we also observed a decrease in S-nitrosylation of tTG and an increase in tTG-mediated cross-linking in the aortas of old versus young rats and humans with little or no changes in the protein expression levels.Citation12 However, the regulation of tTG by NO does not stop here.
NO has recently been shown to alter tTG subcellular distribution and inhibit its deposition into the ECM of fibroblasts.Citation20 Given the abundance and prominent externalization of tTG in the vasculatureCitation1,Citation2,Citation12 and the impact of NO on protein trafficking and secretion in blood vessels,Citation13,Citation14 we sought to determine its effects on tTG localization and functions in vascular cells. Using cell surface biotinylation and immunofluorescence we observed that endogenous NO alters the subcellular distribution of tTG by increasing its levels on the surface and in the ECM of HAECs (data not shown). This was also the case with exogenous or endothelial-derived NO and localization of tTG in human aortic smooth muscle cells (HASMCs)(data not shown).Importantly, this NO-dependent regulation of tTG localization was also observed ex vivo when we found that reducing NO bioavailability by endothelial denudation or inhibition of nitric oxide synthase (NOS) and in vivo in the aortas of old versus young rats significantly increases the levels of ECM-associated tTG (data not shown).
To examine whether this regulation occurs on the level of tTG secretion to the cell surface, we defined the effects of NO on externalization of de novo synthesized protein in HAECs (). The cells were metabolically labeled, cell surface biotinylated and the amounts of de novo synthesized tTG in the fraction of biotinylated (cell surface) proteins were defined by immunoprecipitation. Analysis of time course of tTG externalization showed that NO donor S-nitrosoglutathione (GSNO) significantly inhibits the secretion of tTG, whereas NOS inhibitor L-nitroarginine methyl ester (L-NAME) increases the amounts of tTG on the surface of HAECs. Pretreatment of cells with endocytic inhibitor dynasore which blocks GTPase activity of dynamins,Citation21 did not alter the levels of surface tTG at early time points of secretion, confirming that NO regulates the rate of tTG delivery to the cell surface (not shown). Therefore, we concluded that both exogenous and endogenously produced NO attenuates the process of tTG secretion in these cells (). This second layer of tTG regulation by NO in the vasculature appears important, because, in addition to inhibition of its cross-linking activity, it decreases extracellular levels of the protein, thereby diminishing its nonenzymatic impact on outside-in signaling by integrins, syndecan-4 and PDGFR.
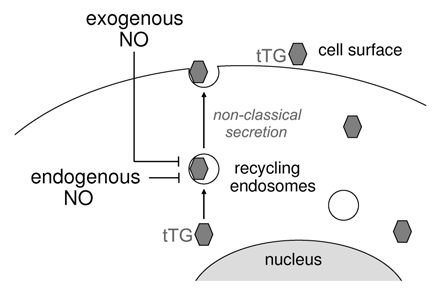
How this regulation may occur? At this point, one can not exclude that S-nitrosylation of tTG itself by NO inhibits its externalization. Mapping and mutating the affected cysteine residues in tTG and analysis of secretion rates of the resulting mutants should address this possibility. However, more likely, NO-dependent modification of NSF, which inhibits its ability to disassemble SNARE complexes, Citation13,Citation14 is pivotal for attenuation of tTG secretion. Specifically, the last step in tTG externalization, a fusion of tTG-bearing recycling endosomes with plasma membraneCitation7 might be impaired by NSF S-nitrosylation. While spatiotemporally controlled reversible NO-mediated regulation of protein trafficking machinery has been attested as a general pathophysiological mechanism in the vasculature, NSF is emerging as its principal redox sensor which impacts not only classical secretion pathways and fusion of Golgi-derived vesicles with plasma membrane, but also unconventional export of cytoplasmic secretory proteins such as tTG.
Figures and Tables
Figure 1 Effects of NO on externalization of endogenous tTG in HAECs. (A) Untreated cells and cells pretreated with NO donor GSNO (200 µm, 1 h) or NOS inhibitor L-NAME (100 µm, 18 h), were metabolically labeled with 35S-translabel for 0–8 h. at the end of labeling, cells were surface-biotinylated with Sulfo-NHS-LC-biotin and cell surface protein fractions were isolated on neutravidinagarose.Citation5,Citation7 the de novo synthesized tTG was immunoprecipitated from total cell extracts (left parts) and cell surface protein fractions (right parts). the resulting tTG immune complexes were resolved by SDS-PAGE and visualized by fluorography. Large and small arrowheads mark the positions of tTG (∼80 kDa) and its major proteolytic fragment (∼68 kDa). (B) the relative amounts of de novo synthesized cell surface tTG were quantified by scintillation counting and compared to those of total tTG. Shown in (A) is a representative of four independent experiments. Bars in (B) depict means ± Sem. *p < 0.05.
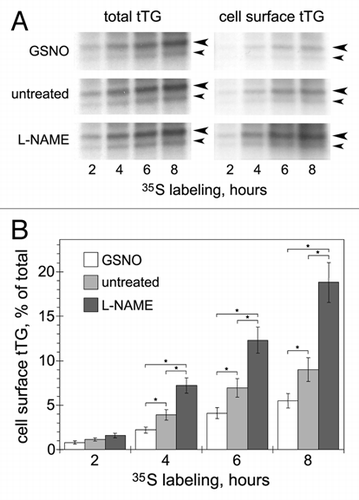
Figure 2 A scheme depicting the effects of exogenous and endogenously produced NO on nonclassical secretion of cytoplasmic tTG.
Acknowledgments
This work was supported by NIH grant 2RO1 HL105296 to D.E.B.
Addendum to:
Related Research Data
References
- Lorand L, Graham RM. Transglutaminases: cross-linking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 2003; 4:140 - 156
- Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 2009; 89:991 - 1023
- Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol 2000; 148:825 - 838
- Telci D, Wang Z, Li X, Verderio EA, Humphries MJ, Baccarini M, et al. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired adhesion through syndecan-4 and beta1 integrin co-signaling. J Biol Chem 2008; 283:20937 - 20947
- Zemskov EA, Loukinova E, Mikhailenko I, Coleman RA, Strickland DK, Belkin AM. Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J Biol Chem 2009; 284:16693 - 16703
- Gentile V, Saydak M, Chiocca EA, Akande O, Birckbichler PJ, Lee KN, et al. Isolation and characterization of cDNA clones to mouse macrophage and human endothelial cell tissue transglutaminases. J Biol Chem 1991; 266:478 - 483
- Zemskov EA, Mikhailenko I, Hsia RC, Zaritskaya L, Belkin AM. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS One 2011; 6:19414
- Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, et al. Small artery remodeling depends on tissue-type transglutaminase. Circ Res 2005; 96:119 - 126
- Johnson KA, Polewski M, Terkeltaub RA. Transglutaminase 2 is central to induction of the arterial calcification program by smooth muscle cells. Circ Res 2008; 102:529 - 537
- Matlung HL, Groen HC, de Vos J, van Walsum T, van der Lugt A, Niessen WJ, et al. Calcification locates to transglutaminases in advanced human atherosclerotic lesions. Am J Pathol 2009; 175:1374 - 1379
- Matlung HL, VanBavel E, van den Akker J, de Vries CJ, Bakker EN. Role of transglutaminases in cuff-induced atherosclerotic lesion formation in femoral arteries of ApoE3 Leiden mice. Atherosclerosis 2010; 213:77 - 84
- Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, et al. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ Res 2010; 107:117 - 125
- Lowenstein CJ. Nitric oxide regulation of protein trafficking in the cardiovascular system. Cardiovasc Res 2007; 75:240 - 246
- Lowenstein CJ, Tsuda H. N-ethylmaleimide-sensitive factor: a redox sensor in exocytosis. Biol Chem 2006; 387:1377 - 1383
- Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 2003; 115:139 - 150
- Morrell CN, Matsushita K, Chiles K, Scharpf RB, Yamakuchi M, Mason RJ, et al. Regulation of platelet granule exocytosis by S-nitrosylation. Proc Natl Acad Sci USA 2005; 102:3782 - 3787
- Ferlito M, Irani K, Faraday N, Lowenstein CJ. Nitric oxide inhibits exocytosis of cytolytic granules from lymphokine-activated killer cells. Proc Natl Acad Sci USA 2006; 103:11689 - 11694
- Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci USA 2006; 103:1295 - 1300
- Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS. Calcium regulates S-nitrosylation, denitrosylation and activity of tissue transglutaminase. Biochemistry 2001; 40:4904 - 4910
- Telci D, Collighan RJ, Basaga H, Griffin M. Increased TG2 expression can result in induction of transforming growth factor beta1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J Biol Chem 2009; 284:29547 - 29558
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 2006; 10:839 - 850