Abstract
The native soluble as well as different aggregated states of recombinant prion proteins are highly sensitive to high pressure. On the one hand, its application to the native α-helical protein induces reversibly a metastable structure that relaxes to amyloid fibrils after prolonged incubation. On the other hand, its application to synthetic prion amyloid fibrils leads to partial disaggregation into native monomers as well as to proto-filaments that have lost several amyloid features. In addition, heat-induced β-sheet prion protein aggregates are dissolved and revert into α-helical monomers by applying high pressure. This profound pressure sensitivity of prion protein structure is explained by large volume differences of the different structural states. Hence, pressure appears as a suitable thermodynamic parameter for exploring the highly complex conformational landscape of prion protein. Its further analysis should help identifying prion protein structural states that are on the pathogenic pathway.
Keywords: :
Alternative folding (misfolding) of prion protein (PrP) emerges as the onset of fascinating cellular events that have challenged some of the most established scientific dogmas. The presently prevalent hypothesis proposes that the resulting abnormal PrP structure (PrPSc), as opposed to the normal cellular PrP structure (PrPC), causes several protein-based neurodegenerative diseases, the so-called prion diseases or transmissible spongiform encephalopathies.Citation1 The two protein isoforms have profoundly different biochemical and biophysical properties. PrPC is soluble in mild detergents and easily degradable by PK, whereas PrPSc is insoluble in mild detergents and highly resistant to PK digestion.Citation2 Furthermore, spectroscopic studies have revealed that the two isoforms have markedly different secondary structures; PrPC consists largely of α-helices, whereas PrPSc is rich in β-sheet structure. The transmission of these disorders occurs either via genetic mutations or by infection. In the latter case, the disease propagation by replication of PrP misfolding is one of the most discussed findings in biological sciences, and is in fact supported by the discovery of prion-like proteins in yeast and fungi that behave as non-mendelian genetic elements.Citation3-Citation5
The general pathology of prion diseases consists in neuronal loss, spongiosis and astrogliosis, accompanied by an accumulation of microglia and, occasionally, the presence of structured PrP deposits, called amyloids.Citation6 Interestingly, several other soluble proteins and polypeptides, with no sequence similarity and structurally different, have been identified to self-associate similarly in such an abnormal manner, and each of them is also related to a particular neurodegenerative or systemic disease, such as Alzheimer disease, Parkinson disease and peripheral amyloidosis.Citation7 Although there are still many caveats regarding the precise structure of amyloids, it is generally considered that all forms of amyloids share the presence of proteolytically resistant and β-structured fibrils. These proteinaceous fibrils are long, straight, and unbranched filaments of 7–12 nm in diameter, with β-strands organized nearly perpendicular to the long axis of the fibril. Amyloid structures have the crystalline property of birefringence and bind to dyes such as Congo red and ThT. There is no specific treatment for amyloidoses, and usually these diseases are fatal.
The ultimate aim of studying the mechanisms of PrP folding is to develop amyloid based therapeutic strategies. Although fundamental differences distinguish de novo protein folding from experimental protein refolding, the availability of in vitro tractable model systems, together with experimental improvement, brought about a noteworthy advance of the knowledge of amyloid formation from a soluble protein. The experimental conditions used to study prion amyloid fibrillogenesis vary within a wide range of parameters. Indeed, the protein structure, and thus the potential energy surface of the protein, has been found to be sensitive to a wide variety of environmental conditions, including pressure, temperature, pH, buffer, ionic strength, as well as the concentration and nature of denaturant agents. Furthermore, the presence of other compounds (ligands) can induce PrP conformational changes. Since amyloidogenesis appears to be controlled by thermodynamic stability and kinetics, destabilizing conditions are used for shifting the dynamic balance between native and unfolded states.Citation8 Usually, this is performed under conditions in which noncovalent interactions still remain favorable, for example by using low concentrations of chaotropic denaturants. Such conditions can play an active role in increasing the roughness of the energy landscape, such that aggregation-prone intermediates can be effectively trapped, by redirecting them toward another folding funnel that leads to a thermodynamically stable fibril conformation. Although these conditions seem far from physiological, they may mimic the in vivo process. Indeed, although the mechanism of conversion from PrPC to PrPSc is still under study, it is assumed that it requires to a certain degree some unfolding of the PrP protein. From these studies it emerges that amyloid fibrils do neither develop directly from the native state of the protein nor from the completely unfolded state, but from intermediate states (only transiently populated), which appear to be partly folded, or to be in a different fold than the native protein. Such intermediates, which have been identified also for many other proteins, are relatively compact with a high content of secondary structure, and may be closely related to normal folding intermediates referred to as ‘molten globule’ states. Although evolution has imposed a short-lived character to these partially folded states, alterations to the protein sequence (mutations) or a malfunction of the cellular protective mechanisms (molecular chaperones and/or ubiquitin proteasome system) can strongly influence their regular population in vivo. Proteins in these conformational states expose hydrophobic amino acid residues to the solvent. Via these exposed hydrophobic surfaces, they tend to associate with one another in a concentration-dependent manner, leading to protein aggregation. Amyloid fibrils appear to be formed via a transitory aggregation nucleus (oligomer), where protein monomers are subsequently captured (in a nucleation-dependent way) and converted by a variety of intermolecular linkages into a fibrillar structure.Citation9,Citation10
The conversion of transiently populated states into aggregated structures (i.e., the digression-point between normal and aberrant folding), the nature of different folding/unfolding events, as well as structural properties between different states are being explored in our laboratory by using high pressure. In this review we present our most relevant results related to the PrP misfolding under pressure. We are focusing on several routes of folding/unfolding that have been unraveled by using pressure.
One of the interests of using high pressure is that it may increase the conformational flexibility of the protein, enabling it to adopt and accumulate in a partially folded state. The propensity of an intermediate conformer to form intermolecular linkages leading to aggregation is a step that is often mediated via hydrophobic and hydrogen bonds interactions by key residues that are usually shielded. However, because inter-protein forces are of the same nature like those involved in correct folding, there is a competition between the correct folding and intermolecular assembling that may depend on the energy as well as on the volume of each particular state involved. Since pressure is the thermodynamic parameter that fits best to exploit differences in volume, we have used pressure as a tool to modulate folding, misfolding and aggregation processes.
Conversion of Soluble Native Protein to Amyloid Fibrils
In a first study,Citation11 restricted to a low PrP concentration (0.5–0.8 mg/mL), we revealed simple two-state, reversible pressure-induced unfolding transitions. The results suggested that even at very high pressure (up to 600 MPa), unfolding of PrP was not complete. Although no pressure-induced protein aggregation was detected, at pressures greater than 450 MPa, we observed a metastable structure, possibly a soluble oligomer, because of its ThT binding capacity.
However, in further studies with higher protein concentrations and more alkaline pH, the metastable state was found to aggregate.Citation12 Indeed, at 2 mg protein/ml and pH 8.5, the protein underwent irreversible aggregation at pressures above 400 MPa. Concomitantly, a gradual and large increase in the fluorescence intensities of ANS and ThT was observed, indicating that the intermediates participating in aggregation were partially unfolded and were possibly of amyloidogenic nature. PrP aggregates recovered after immediate decompression showed an increase in β-sheet content and an acquired resistance to proteolysis (resistance to PK).
Subsequently, we observed that depending on the incubation time under high pressure (600 MPa), pre-amyloid structures or mature amyloid fibrils could be obtained.Citation12 The pre-amyloid structures (formed after a very short pressure incubation time) were small globular aggregates, that bound ThT and ANS. Their secondary structure was rich in β-sheet content. In contrast, aggregates formed after prolonged incubation under high pressure (see for a schematic view) showed a typical amyloid fibrillar structure when observed by electron microscopy. These fibrils, which coexisted with non-amyloid aggregates, were capable of ThT and Congo red binding with the associated green birefringence under polarized light. In addition, these fibrils showed a strong β-sheet content, and they were resistant to PK digestion. In contrast to the fibrillar PrP forms described in previous reports that had been formed by traditional approaches using chemical denaturants, the amyloid fibrils formed under pressure do not bind ANS. The incapacity of these fibrils to bind ANS may be explained by an occlusion of the ANS binding sites (exposed hydrophobic regions) due to unique intermolecular contacts within the fibrillar network. This result suggests that high pressure induces an alternative misfolding pathway leading to a structurally distinct amyloid, probably by selecting conformational changes accompanied by a decrease in volume.
Figure 1. Schematic representation of the effects of high pressure to native soluble as well as different aggregated states of recombinant prion proteins.
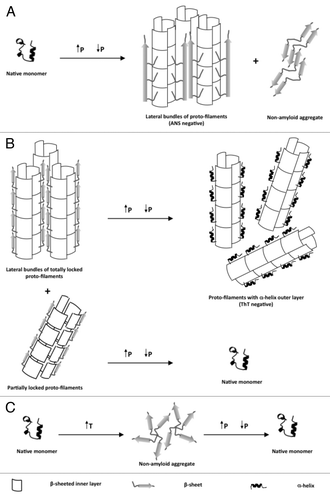
The interplay of different parameters, such as pressure, temperature, pH, presence of co-factors, and incubation time to the nature of the macroscopic PrP structural changes have been further studied by our group in a number of subsequent works,Citation13,Citation14 which confirmed the unusually strong dependence of the final structural state on small differences of physico-chemical parameters.
Conversion of Amyloid Fibrils into Non-amyloid Fibrils and Soluble Monomers
The combined effects of high pressure and temperature on the infectivity titer in processed meat contaminated by prions have been investigated.Citation15,Citation16 In particular, Fernandez Garcia et al.Citation16 showed a remarkable efficiency of pressures higher than 500 MPa combined to temperature of only 60°C in decreasing the resistance of PrPSc to PK. A significant delay of the onset of disease in animals infected with the pressurized material was also observed. These irreversible effects are certainly due to a pressure induced destabilization of PrPSc. Since PrP from scrapie infected brains is isolated as amyloid fibrils, the general assumption is that infectious PrPSc is made up of amyloid fibrils. This would mean that PrP amyloid fibrils dissociate under pressure - at least to some extent.
Indeed, high pressure can also be used to dissolve PrP amyloid fibrils.Citation17 This is schematized in . We showed the nearly complete and irreversible loss of ThT binding to amyloid fibrils. In this case, the starting PrP structural state was mature amyloid fibrils that had been prepared by the protocol of Baskakov.Citation8,Citation18 The decrease of fluorescence of ThT upon fibril dissociation has then been used to analyze the kinetics of the pressure-induced amyloid fibril dissociation. The dissociation process was biphasic, and the kinetic rates were strongly pressure and temperature dependent. Depending on pressure and temperature, the dissociation occurred within minutes to hours. However, the pressure-induced dissociation was not complete. The remaining fibrils were composed of a significant amount of shortened and thinned fibrils.
The fact that not all fibrils were dissociated by pressure is intriguing. Analysis by complementary techniques, such as fourier transform infrared spectroscopy, fluorescence, electrophoresis and toxicity assays indicated an inhomogeneity of the starting amyloid fibrils. Part of them consisted of very stable, termed “totally locked” and others of rather labile, termed “partially locked” protofilaments. Application of pressure to the totally locked form produced protofilaments that had lost part of their amyloid characteristics. In contrast, application of pressure to the partially locked form led to their complete dissolution. Both pathways can be seen as proceeding via a mechanistically similar unfolding process that implies a disordered and hydrated kinetic transition state.
Conversion of heat-induced β-sheet aggregates into α-helical monomers
Not only high pressure but also heat can be used to transform the soluble and mainly α-helical structure of PrPC into insoluble aggregates that are rich in intermolecular β-sheet structure. However, heat-induced are different from pressure-induced aggregates. They are amorphous – i.e., non-amyloid. Furthermore, the thermodynamics of pressure and heat induced structural changes were different.Citation11,Citation19 As depicted in , upon increasing pressure (200 MPa), the temperature-induced aggregates dissolved, and once the pressure was released the protein refolded to its native α-helical secondary structure. The kinetics of their dissolution occurred within a fraction of seconds, which was must faster than the pressure-induced dissociation of amyloid fibrils.
Several reasons may explain why pressure can prevent (or reverse) protein aggregation. The most obvious one is the capacity of pressure of hydrating hydrophobic residues that become solvent exposed upon heating, thus preventing their intermolecular interactions. Other possible explanations of pressure dissociation of protein aggregates include: (1) the existence of small “free volumes” (packing defects) at the intermolecular boundaries in aggregates. Upon pressure-induced dissociation the solvent molecules can be packed much better on the surface of the protein interfaces; (2) the hydration of dissociated protein-protein salt linkages (electrostriction effect).
Conclusions
Many researchers are looking for the “missing state” in the sequence of events leading to the malignant prion form. The results presented here suggest that this precursor state can be reached by applying high pressure to either the native or the fibrillar prion form. This state appears to be highly instable – it can easily transform into either monomeric or fibrillar forms under small changes of the environment. Pressure appears to be particularly useful for imposing the direction and the final state of these structural transformations. Now, pressure effects on equilibria and kinetics can be observed only when the initial and final or transition states differ in volume. Hence, the reported strong pressure dependence of the structural transformations must reflect large volume differences of the species involved. These are probably due to strongly varying packing and hydration properties. On the basis of these considerations, one might envisage molecular modeling of the pressure-induced states. This could be interesting for the design of anti prion drugs.
| Abbreviations: | ||
| PrP | = | prion protein |
| ANS | = | 8-anilino-1-naphthalene sulfonate |
| ThT | = | thioflavin T |
| PK | = | proteinase K |
| MPa | = | megapascals |
References
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem 1998; 67:793 - 819; http://dx.doi.org/10.1146/annurev.biochem.67.1.793; PMID: 9759504
- Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA 1997; 94:9773 - 8; http://dx.doi.org/10.1073/pnas.94.18.9773; PMID: 9275200
- Serio TR, Lindquist SL. [PSI+]: an epigenetic modulator of translation termination efficiency. Annu Rev Cell Dev Biol 1999; 15:661 - 703; http://dx.doi.org/10.1146/annurev.cellbio.15.1.661; PMID: 10611975
- Wickner RB, Taylor KL, Edskes HK, Maddelein ML, Moriyama H, Roberts BT. Prions of yeast as heritable amyloidoses. J Struct Biol 2000; 130:310 - 22; http://dx.doi.org/10.1006/jsbi.2000.4250; PMID: 10940235
- Prusiner SB, DeArmond SJ. Prion diseases of the central nervous system. Monogr Pathol 1990; 32:86 - 122; PMID: 2192281
- Carrell RW, Lomas DA. Conformational disease. Lancet 1997; 350:134 - 8; http://dx.doi.org/10.1016/S0140-6736(97)02073-4; PMID: 9228977
- Breydo L, Makarava N, Baskakov IV. Methods for conversion of prion protein into amyloid fibrils. Methods Mol Biol 2008; 459:105 - 15; http://dx.doi.org/10.1007/978-1-59745-234-2_8; PMID: 18576151
- Come JH, Fraser PE, Lansbury PT Jr.. A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc Natl Acad Sci USA 1993; 90:5959 - 63; http://dx.doi.org/10.1073/pnas.90.13.5959; PMID: 8327467
- Jarrett JT, Lansbury PT Jr.. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie?. Cell 1993; 73:1055 - 8; http://dx.doi.org/10.1016/0092-8674(93)90635-4; PMID: 8513491
- Torrent J, Alvarez-Martinez MT, Heitz F, Liautard JP, Balny C, Lange R. Alternative prion structural changes revealed by high pressure. Biochemistry 2003; 42:1318 - 25; http://dx.doi.org/10.1021/bi0269916; PMID: 12564935
- Torrent J, Alvarez-Martinez MT, Harricane MC, Heitz F, Liautard JP, Balny C, et al. High pressure induces scrapie-like prion protein misfolding and amyloid fibril formation. Biochemistry 2004; 43:7162 - 70; http://dx.doi.org/10.1021/bi049939d; PMID: 15170353
- El Moustaine D, Perrier V, Smeller L, Lange R, Torrent J. Full-length prion protein aggregates to amyloid fibrils and spherical particles by distinct pathways. FEBS J 2008; 275:2021 - 31; http://dx.doi.org/10.1111/j.1742-4658.2008.06356.x; PMID: 18355314
- El Moustaine D, Torrent J, Lange R. Prion protein aggregation induced by copper (II) and heparan sulfate. Pressure dependent switch of reaction pathways. Z Naturforsch 2008; 63b:745 - 55
- Brown P, Meyer R, Cardone F, Pocchiari M. Ultra-high-pressure inactivation of prion infectivity in processed meat: a practical method to prevent human infection. Proc Natl Acad Sci USA 2003; 100:6093 - 7; http://dx.doi.org/10.1073/pnas.1031826100; PMID: 12732724
- Fernández García A, Heindl P, Voigt H, Buttner M, Wienhold D, Butz P, et al. Reduced proteinase K resistance and infectivity of prions after pressure treatment at 60 degrees C. J Gen Virol 2004; 85:261 - 4; http://dx.doi.org/10.1099/vir.0.19410-0; PMID: 14718641
- El Moustaine D, Perrier V, Acquatella-Tran Van Ba I, Meersman F, Ostapchenko VG, Baskakov IV, et al. Amyloid features and neuronal toxicity of mature prion fibrils are highly sensitive to high pressure. J Biol Chem 2011; 286:13448 - 59; http://dx.doi.org/10.1074/jbc.M110.192872; PMID: 21357423
- Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease. Protein Sci 2005; 14:1222 - 32; http://dx.doi.org/10.1110/ps.041186605; PMID: 15802644
- Torrent J, Alvarez-Martinez MT, Liautard JP, Balny C, Lange R. The role of the 132-160 region in prion protein conformational transitions. Protein Sci 2005; 14:956 - 67; http://dx.doi.org/10.1110/ps.04989405; PMID: 15772306