Abstract
The main energetic resources of the cell are the mitochondria. As such, these organelles control a number of processes related to the life and death of the cell and also have a prominent function in the maintenance of tumor cells. In the last years, several authors have proposed an active role for mitochondria in tumorigenesis, more specifically concerning somatic mutations in mitochondrial DNA (mtDNA). Here, we wanted to evaluate this hypothesis based on the conclusions obtained in a model of gliomagenesis with elevated levels of ROS (reactive oxygen species), a toxic by-product of tumor metabolism. According to our findings, none of the mtDNA variants were found relevant to the tumoral process or suggest the involvement of mitochondria in tumorigenesis beyond the metabolic requirements of the tumoral cell. We conclude that there is not enough evidence to support the claim that mitochondrial instability holds any relevant role in the tumoral process.
Mitochondria are involved in numerous and important processes such as the homeostatic control of calcium, iron and heme biogenesis.Citation1,Citation2 They also control certain kind of programmed cell death or apoptosis by releasing cytochrome c and other factors into the cytosol that trigger the apoptotic intrinsic pathway.Citation3 However, mitochondria are best known as critical players in regulating different stages of cellular respiration such as terminal electron transport, the citric acid cycle, and oxidative phosphorylation. Along this process, mitochondria use metabolic intermediates generated during the tricarboxilic acid (TCA) cycle to generate ATP that will be subsequently employed in other biosynthetic reactions.Citation4
An alternative pathway of energy production is the conversion of glucose to lactic acid in a process known as ‘aerobic glycolysis’, which in mammalian cells is usually inhibited by the presence of O2. This inhibition grants the cell a wide range of oxygen concentrations under it is possible to maintain the energy production. As Otto Warburg noticed in 1926,Citation5 this type of glycolysis is a hallmark of cancer cells, which heavily rely on glycolysis rather than cellular respiration to produce most of their ATP even when O2 is plentiful.
The rapid and chaotic growth of most tumor types entails a hypoxic environment that facilitates the production of reactive oxygen species (ROS), a toxic by-product of the molecular oxygen consumed during respiration.Citation6 Although low levels of ROS are easily manageable by an inducible antioxidant program, an imbalance that favor ROS production can lead to oxidative stress, increased genomic instability and impairment of DNA repair mechanisms that can result in mutations or double strand breaks (DSBs), specially in the mtDNA.Citation7 This adds up to its close proximity to the electron transport chain, the fact that unlike nuclear DNA mtDNA lacks the protective activity of histones and the special susceptibility of mtDNA to organic compounds with carcinogenic activity.Citation8 All of this makes the mtDNA of tumor cells a suitable candidate to harbor somatic mutations that may be responsible for malignant transformation.
In this mini-review we want to extent on our results and conclusions regarding our recent publication in PLoS ONE,Citation9 where we found that although ROS may have a prominent role in early tumorigenesis helping the tumor cell to gain new oncogenic hits, this is solely true in somatic mutations affecting the nuclear DNA, while mtDNA remained intact along the process.
Mitochondrial dysfunctions have long been reported and were also hypothesized to contribute to several stages related to tumorigenesis. Although the mechanisms by which this contribution takes place are still unclear, several authors have described mtDNA mutations as driving force promoting tumoral growth,Citation10 malignancyCitation11 or modulating its metastatic potential.Citation12,Citation13 By contrast, other authors question the validity of these findings arguing that may be explainable by errors in documentation or laboratory work.Citation14,Citation15
Several examples of the occurrence and involvement of mutated mtDNA in carcinogenesis are reported by Ishikawa and colleagues.Citation12 The authors examined the contribution of mtDNA mutations in several cell lines with different metastatic potential by performing reciprocal exchange of mitochondria among them, thus creating transmitocondrial cybrids. These cybrids from cell lines, either of murine or human origin, displayed switched mitochondrial activities, ROS levels and also metastatic potential when injected in C57BL/6 mice. To verify whether these findings have a correlate with mtDNA integrity, the authors analyzed the mtDNA of highly metastatic cell lines and identified two mutations in these cells as the responsible for mitochondrial dysfunction. Accordingly, mutated mtDNA from highly metastatic cell lines, together with defects in complex I, upregulates transcription factors associated with the metastatic phenotype such as MCL-1, HIF-1α and VEGF. This metastatic behavior seems to be highly dependent on the integrity of mtDNA, as these same authors propose in another recent communication.Citation13 In this study, mitochondria from MDA-MB 231 cells were replaced with mitochondria carrying normal mtDNA. Although ROS levels did not decrease the impaired mitochondrial respiratory function of this breast cancer cell line was successfully recovered and its metastatic potential suppressed. Finally, they suggest that although either nuclear and mitochondrial DNA share the control over the respiratory function of the cell, only defects in mtDNA confer the cell the ability to metastasize in the absence of ROS production.
High levels of ROS are a common feature among several human cancer cell lines and tumors from different lineages. Several oncogenes such as H-RasV12, K-RasG12D and c-Myc are known to increase these intracellular levels of ROS.Citation16 In our model of gliomagenesis,Citation17 we combined two oncogenic hits, H-RasV12 and deletion of retinoblastoma (Rb1), to recapitulate two common features of gliomas, hyper-activation of signaling pathways and deregulation of cell cycle control, respectively.Citation18 We observed that although astrocytes with high levels of ROS produced by H-RasV12 underwent cellular transformation they only displayed features of malignization when combined with retinoblastoma loss. These cells were subsequently injected in SCID immunodeficient mice and the tumoral masses derived were analyzed. We found that tumors from H-RasV12 expressing astrocytes displayed similarities to low-grade human gliomas, while tumors obtained from astrocytes with both hits resembled high-grade gliomas. Interestingly, we found that H-RasV12 alone was sufficient to induce high levels of ROS and chromosomal instability, whereas the contribution of Rb deletion was not significant.
In order to analyze the involvement of mtDNA in tumoral progression we also compared the karyotypes of cells derived from cRbloxP/loxP/H-RasV12 and cRb−/−/RasV12 tumors, T653 (cRbloxP/loxP/H-RasV12) and T731 (cRb−/−/RasV12) respectively, and also with that of cRbloxP/loxP/H-RasV12 and cRb−/−/RasV12 primary astrocytes. Curiously, we found that although in primary astrocytes the replication stress induced by the oncogene H-RasV12 alone is able to produce genetic aberrations in 30% of the cells, the cell lines showed severe aberrations and duplications of the chromosomes, ranging from 60 to > 80 in most cases, in virtually 100% of the population analyzed. This seems to not be the case with mtDNA; although mitochondria are more likely to be targeted by oxidizing agents no significant instabilities were found. In the same way, analysis of human glioma samples mirrored previous results given that none of the variants found were suspected to be involved in the cancerous process.
These data indicates that cells with the greater number of aberrations were positively selected probably due to additional hits gained that somewhat allowed for maintenance and progression of tumorigenesis. Recently, DeNicola et al.Citation19 have suggested that Nrf2 (also known as nuclear factor erythroid 2-related factor) activity is linked to RAS-mediated ROS production through the RAF-MEK-ERK pathway. This factor, along with its repressor KEAP1, regulates the expression of numerous detoxifying and antioxidant genes, serving as a protection against carcinogenesis, liver toxicity or respiratory distress.Citation20 The authors also found decreased ROS levels in pancreatic and lung cancer samples from patients and mice, coincidentally with high levels of NRF2 and its target genes. However, elimination of NRF2 in these types of tumors increased the number of senescent cells and reduced the proliferative rate of K-RasG12D, suggesting that tumoral cells may take advantage of this ROS-detoxifying pathway to enhance survival.
Cytochrome c oxidase activity is a good indicator of the respiratory activity of the mitochondrion since this is a transmembrane protein that serves as terminal electron acceptor in the electron transfer chain. Intriguingly, the mitochondrial activity in transduced astrocytes was considerably higher than that of tumor cells, who appeared on par to control groups. A similar outcome was observed with ROS levels, higher production in cRbloxP/loxP/H-RasV12 and cRb−/−/H-RasV12 primary astrocytes but lower in the cell lines, suggesting that the primary source of these ROS was of mitochondrial origin. Likewise, the quantity and distribution of the mitochondria within the cell seemed to corroborate these data.
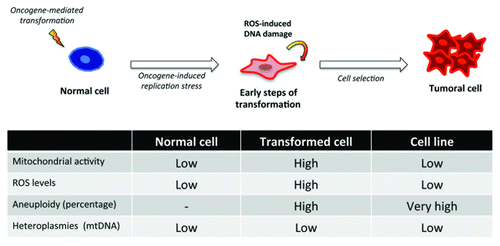
Taken together, and as shows, these data suggested that the hyper-activation of signaling pathways mediated by oncogenic stimuli as RAS leads to a DNA replicative stress that in some cell types is thwarted by the cell entering senescence,Citation21 but not in the case of astrocytes.Citation17 In this initial stage, RAS-expressing cells are highly mutagenic either because of this oncogene-induced replicative stress or impairments in the DNA repair response. This stress also affects the mitochondria, which are over-stimulated to reach the metabolic needs of the transforming cell, favoring the production of highly reactive oxygen species that in turn mutate nuclear DNA promoting carcinogenesis. However, and despite other authors suggesting opposite scenarios,Citation22 mtDNA remains without any noticeable or significant mutation that could contribute to this process. Curiously, these transformed cells seem to undergo a selection process where the cells with the most genetic aberrations pass, probably due to the activation of detoxification programs that help the cell reduce toxic ROS levels.Citation19
Figure 1. In our experimental setting a ‘normal cell’, an astrocyte is submitted to an oncogenic event, H-RASV12 that induces an oncogene-mediated transformation by means of the hyperactivation of the signaling pathways, and very especially the RAF-MEK-ERK pathway. This causes a replication stress that in other cell types leads to senescence but in astrocytes increases their proliferation rate, transforming the cell in the process. The reactive oxygen species present in such scenario allow for additional oncogenic insults that help the transforming cell complete the process, involving a positive selection of cells with the greater number of genetic aberrations in nuclear DNA. Surprisingly, none of the heteroplasmies found in the mitochondrial DNA of tumor cells were linked to the onset or maintenance of the tumoral process.
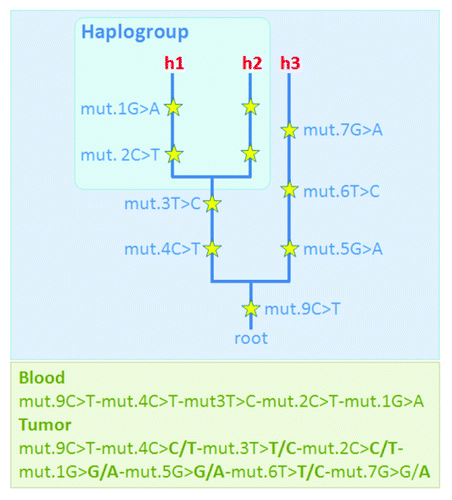
Methodological errors occur in all facets of genetic research, including mtDNA studies. Salas et al.Citation15 indicated that “the vast majority (80%) of the studies dealing with potential functional implications of the mtDNA molecule in tumorigenesis (and providing data for inspection) are based on faulty data with surreal findings.” These conclusions were based on solid theoretical foundations.Citation23 Briefly, the mtDNA is inherited as a single block from the mother to the offspring such that inter-generational mtDNA differences only arise by way of mutation. This feature, couple with the fact that there is a huge amount of genomic data available in the literature (> 150,000 control region sequences and > 9,500 entire genomes analyzed in hundreds of human population groups) has allowed the reconstruction of the most solid phylogeny in the whole field of genetics. Thus, an experienced researcher can easily detect artificial patterns of mtDNA variation (generated by errors) when contrasting these patterns against those expected according to the known mtDNA tree. For instance, the instabilities detected by oncogeneticists can often be most plausibly explained by way of unintended mixtures of two or more lineages representing different branches of the mtDNA phylogeny, one could be the genuine one while the other from a contaminant (). In such cases, it is almost impossible to conceive such mixture scenario by the action of the tumor cell machinery.Citation24 Therefore, in tumor studies, seeming instabilities can easily occur by e.g., contamination, sample mix-up, phantom mutations or documentation errors, given the fact that quite often low amount DNA are manipulated in these studies and micro-dissection experiments are performed without proper quality standards.
Figure 2. Scheme showing a case were a tumor sample shows several ‘seeming-instabilities’ when compared with the blood (non-tumor) sample from the same patient; instead of attributing these (seeming) instabilities to the tumor process, one could considers an alternative more likely explanation. Thus, one could assume the occurrence of an unfortunate contamination of the tumor sample, and therefore the observed tumor profile could be better explained as a mixture of two haplotypes (h1+h3): one haplotype corresponds to the genuine one from the patient (h1) and the other (h3) comes from an external donor (contaminant); the phylogenetic approach allows to easily deconvolute the mixture. Along branches of the fictitious phylogeny are the mutations (yellow starts), beside are the numbers indicating the mutational positions and the letters (A, C, G, T) indicating the nucleotide variant; e.g mut.1G > G/A means that at position 1, the reference sequence has a G, while the targeted haplotype carries two variants simultaneously, G and an A. More about notation: a) haplotypes are annotated from the root (the reference sample) to the tip of the branches); b) “h” denotes haplotypes (#1, #2 and #3); and c) haplogroup is a clade of sequences phylogenetically related, in this particular example, haplotypes 1 and 2 belong to the same haplogroup because they share the diagnostic mutations (basal motif) at positions 3 and 4.
The so-popularized phylogenetic approach has resulted to be extremely useful in other fields of research, such as population genetics; and although guidelines have been published in order to minimize errors in genetic studies,Citation25,Citation26 the medical literature still shows a high penetrance of errors of different nature.Citation27-Citation30 The interest of forensic analysts in the mtDNA test, especially for the analysis of hair shafts and skeletal remains, has obligated them to pay deep attention to the different source of errors in mtDNA analysis;Citation31-Citation34 and this has stimulated them to substantially improve quality standards.
Unfortunately, errors still exist in the literature dealing with mtDNA instability.Citation35,Citation36 This is favored by the fact that studies presenting a good number of instabilities find their way into oncogenetic journals much easier than those that just show negative results (a problem that has been baptized in biomedical sciences as ‘publication bias’). Accepting studies that do not follow minimal quality —e.g. ‘forensic’— standards (as for instance performed in Cerezo et al.Citation36) will unfortunately contribute to perpetuate a fictitious scenario that drives science in wrong directions. A similar scenario is that involving case-control mtDNA association studies.Citation37,Citation38
It is most likely that the power of next generation sequencing (NGS) techniques will help to unravel the role (if any) that mtDNA variation could have in tumorigenesis. However, before NGS replace standard Sanger sequencing procedures, it needs proper validation, preferably following the high standards demanded by forensic geneticists.Citation39
In summary, we have demonstrated that in our model of gliomagenesis numerous and important alterations of nuclear DNA. Mitochondrial activity and ROS production is highly increased in these early steps of tumorigenesis but mtDNA conserved its integrity along the process. Thus, the possibility of intervening in mitochondrial energy metabolism function in cancer is therapeutically promising, although up to now, the number of molecules available with potential to safely achieve this is limited.Citation40 These data were corroborated with human samples of patients with glioma, thus ruling out a mitochondrial genetic instability contribution to tumorigenesis in this type of cancers. There are no guaranties for the findings presented by previous studies indicating a number of mtDNA instabilities as responsible for tumorigenesis, under the suspicion that these results could be false positives of instabilities artificially and unintentionally generated by different kind of methodological errors.
| Abbreviations: | ||
| mtDNA | = | mitochondrial DNA |
| ROS | = | reactive oxygen species |
| TCA | = | tricarboxilic acid |
| DSBs | = | double strand breaks |
| Rb1 | = | retinoblastoma |
Acknowledgments
We thank the members and former members of the Molecular Oncology Laboratory MOL, especially Marcos Seoane, for helpful discussions. This study was supported by the Spanish Ministry of Education and Science SAF2008–00543 (JAC), SAF2009–08629 (JAC), SAF2008–02971 (AS), Xunta de Galicia INCITE08PXIB208091PR (JAC) and Fundación de Investigación Medica Mutua Madrileña 2008/CL444 (AS).
References
- Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: The calcium connection. Biochim Biophys Acta 2010; 1797:607-18.
- Schultz IJ, Chen C, Paw BH, Hamza I. Iron and porphyrin trafficking in heme biogenesis. J Biol Chem 2010; 285:26753 - 9; http://dx.doi.org/10.1074/jbc.R110.119503; PMID: 20522548
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116:205 - 19; http://dx.doi.org/10.1016/S0092-8674(04)00046-7; PMID: 14744432
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 5th Ed. New York: Garland Science, 2007.
- Warburg O. Über den Stoffwechsel der Tumore. Berlin: Springer, 1926.
- Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res 2011; 711:193 - 201; http://dx.doi.org/10.1016/j.mrfmmm.2010.12.016; PMID: 21216256
- Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them?. Trends Cell Biol 2008; 18:165 - 73; http://dx.doi.org/10.1016/j.tcb.2008.01.006; PMID: 18296052
- Allen JA, Coombs MM. Covalent binding of polycyclic aromatic compounds to mitochondrial and nuclear DNA. Nature 1980; 287:244 - 5; http://dx.doi.org/10.1038/287244a0; PMID: 7432460
- Seoane M, Mosquera-Miguel A, Gonzalez T, Fraga M, Salas A, Costoya JA. The mitochondrial genome Is a “genetic sanctuary” during the oncogenic process. PLoS ONE 2011; 6:e23327; http://dx.doi.org/10.1371/journal.pone.0023327; PMID: 21858071
- Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res 2008; 68:700 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-07-5532; PMID: 18245469
- Nishikawa M, Nishiguchi S, Shiomi S, Tamori A, Koh N. Somatic mutation of mitochondrial DNA in cancerous and noncancerous liver tissue in individuals with hepatocellular carcinoma. Cancer Res 2001; 61:1843 - 5; PMID: 11280735
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008; 320:661 - 4; http://dx.doi.org/10.1126/science.1156906; PMID: 18388260
- Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, Takenaga K, et al. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011; 6:e23401; http://dx.doi.org/10.1371/journal.pone.0023401; PMID: 21853128
- Vega A, Salas A, Gamborino E, Sobrido MJ, Macaulay V, Carracedo Á. mtDNA mutations in tumors of the central nervous system reflect the neutral evolution of mtDNA in populations. Oncogene 2004; 23:1314 - 20; http://dx.doi.org/10.1038/sj.onc.1207214; PMID: 14647457
- Salas A, Yao YG, Macaulay V, Vega A, Carracedo Á, Bandelt HJ. A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med 2005; 2:e296; http://dx.doi.org/10.1371/journal.pmed.0020296; PMID: 16187796
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 1999; 274:7936 - 40; http://dx.doi.org/10.1074/jbc.274.12.7936; PMID: 10075689
- Seoane M, Iglesias P, Gonzalez T, Dominguez F, Fraga M, Aliste C, et al. Retinoblastoma loss modulates DNA damage response favoring tumor progression. PLoS ONE 2008; 3:e3632; http://dx.doi.org/10.1371/journal.pone.0003632; PMID: 18985151
- Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2001; 2:120 - 9; http://dx.doi.org/10.1038/35052535; PMID: 11253051
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011; 475:106 - 9; http://dx.doi.org/10.1038/nature10189; PMID: 21734707
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 2004; 10:549 - 57; http://dx.doi.org/10.1016/j.molmed.2004.09.003; PMID: 15519281
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593 - 602; http://dx.doi.org/10.1016/S0092-8674(00)81902-9; PMID: 9054499
- Park JS, Sharma LK, Li H, Xiang R, Holstein D. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum Mol Genet 2009; 18:1578 - 89; http://dx.doi.org/10.1093/hmg/ddp069; PMID: 19208652
- Bandelt HJ, Quintana-Murci L, Salas A, Macaulay V. The fingerprint of phantom mutations in mitochondrial DNA data. Am J Hum Genet 2002; 71:1150 - 60; http://dx.doi.org/10.1086/344397; PMID: 12384858
- Egeland T, Salas A. A statistical framework for the interpretation of mtDNA mixtures: forensic and medical applications. PLoS ONE submitted PMID: 19098988
- Salas A, Carracedo Á, Macaulay V, Richards M, Bandelt HJ. A practical guide to mitochondrial DNA error prevention in clinical, forensic, and population genetics. Biochem Biophys Res Commun 2005; 335:891 - 9; http://dx.doi.org/10.1016/j.bbrc.2005.07.161; PMID: 16102729
- Bandelt HJ, Achilli A, Kong QP, Salas A, Lutz-Bonengel S, Sun C, et al. Low “penetrance” of phylogenetic knowledge in mitochondrial disease studies. Biochem Biophys Res Commun 2005; 333:122 - 30; http://dx.doi.org/10.1016/j.bbrc.2005.04.055; PMID: 15958208
- Bandelt HJ, Salas A, Taylor RW, Yao YG. The exaggerated status of “novel” and “pathogenic” mtDNA sequence variants due to inadequate database searches. Hum Mutat 2009; 30:191 - 6; http://dx.doi.org/10.1002/humu.20846; PMID: 18800376
- Bandelt HJ, Salas A. Contamination and sample mix-up can best explain some patterns of mtDNA instabilities in buccal cells and oral squamous cell carcinoma. BMC Cancer 2009; 9:113; http://dx.doi.org/10.1186/1471-2407-9-113; PMID: 19371404
- Bandelt HJ, Yao YG, Salas A, Kivisild T, Bravi CM. High penetrance of sequencing errors and interpretative shortcomings in mtDNA sequence analysis of LHON patients. Biochem Biophys Res Commun 2007; 352:283 - 91; http://dx.doi.org/10.1016/j.bbrc.2006.10.131; PMID: 17123466
- Bandelt HJ, Salas A, Bravi CM. What is a 'novel' mtDNA mutation—and does 'novelty' really matter?. J Hum Genet 2006; 51:1073 - 82; http://dx.doi.org/10.1007/s10038-006-0066-5; PMID: 17021933
- Bandelt HJ, Salas A, Bravi CM. Problems in FBI mtDNA database. Science 2004; 305:1402 - 4; http://dx.doi.org/10.1126/science.305.5689.1402b; PMID: 15353782
- Bandelt HJ, Salas A, Lutz-Bonengel S. Artificial recombination in forensic mtDNA population databases. Int J Legal Med 2004; 118:267 - 73; http://dx.doi.org/10.1007/s00414-004-0455-2; PMID: 15257464
- Salas A, Bandelt HJ, Macaulay V, Richards MB. Phylogeographic investigations: the role of trees in forensic genetics. Forensic Sci Int 2007; 168:1 - 13; http://dx.doi.org/10.1016/j.forsciint.2006.05.037; PMID: 16814504
- Salas A, Prieto L, Montesino M, Albarrán C, Arroyo E, Paredes-Herrera MR, et al. Mitochondrial DNA error prophylaxis: assessing the causes of errors in the GEP'02-03 proficiency testing trial. Forensic Sci Int 2005; 148:191 - 8; http://dx.doi.org/10.1016/j.forsciint.2004.06.008; PMID: 15639614
- Bandelt HJ, Salas A. Contamination and sample mix-up can best explain some patterns of mtDNA instabilities in buccal cells and oral squamous cell carcinoma. BMC Cancer 2009; 9:113; http://dx.doi.org/10.1186/1471-2407-9-113; PMID: 19371404
- Cerezo M, Bandelt HJ, Martin-Guerrero I, Ardanaz M, Vega A, Carracedo Á, et al. High mitochondrial DNA stability in B-cell chronic lymphocytic leukemia. PLoS ONE 2009; 4:e7902; http://dx.doi.org/10.1371/journal.pone.0007902; PMID: 19924307
- Salas A, Fachal L, Marcos-Alonso S, Vega A, Martinón-Torres F, Esigem G. Investigating the role of mitochondrial haplogroups in genetic predisposition to meningococcal disease. PLoS ONE 2009; 4:e8347; http://dx.doi.org/10.1371/journal.pone.0008347; PMID: 20019817
- Mosquera-Miguel A, Álvarez-Iglesias V, Vega A, Milne R, Cabrera de León A, Benitez J, et al. Is mitochondrial DNA variation associated with sporadic breast cancer risk?. Cancer Res 2008; 68:623 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-07-2385; PMID: 18199560
- Bandelt HJ, Salas A. Current Next Generation Sequencing technology may not meet forensic standards. Forensic Sci Int Genet 2011; In press http://dx.doi.org/10.1016/j.fsigen.2011.04.004; PMID: 21565569
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol 2010; 28:249 - 55; PMID: 20160716