Abstract
Peripheral neuropathies such as Charcot-Marie-Tooth disease (CMT) are a group of neurological disorders that affect the peripheral nervous system. Although demyelinating CMT is the most prevalent hereditary peripheral neuropathy, there are currently no effective treatments for patients suffering from this disease. Recent studies by our group and others have provided a link between protein misfolding and demyelinating CMT and indicate that impairment of the proteasome and aggresome-autophagy pathways may contribute to CMT pathogenesis. These studies suggest that targeting protein quality control systems involved in cytoprotection against CMT-associated misfolded proteins could have therapeutic benefits for treating demyelinating CMT.
Peripheral neuropathies are a group of disorders that affect the peripheral nervous system, with demyelinating Charcot-Marie-Tooth disease (CMT) being the most common inherited peripheral neuropathy.Citation1 Demyelinating CMT is characterized by motor weakness, sensory loss, and muscle wasting, all of which drastically decrease the quality of life.Citation2 There are currently no effective treatments for demyelinating CMT. Recent studies by our laboratory and others have provided important insights into the molecular and cellular pathways involved in demyelinating CMT, which may help develop better treatments for this disease.
Protein Misfolding is a Common Feature in Demyelinating CMT
Our recent study revealed that mutations causing demyelinating CMT type 1C (CMT1C) are clustered within or around the transmembrane domain of the C-tail anchored protein SIMPLE and disrupts its insertion into the membrane, leading to accumulation of misfolded SIMPLE in the cytosol.Citation3 Similarly, mutations causing the overexpression or single amino acid substitutions of the type III tetra-spanningCitation4 membrane protein peripheral myelin protein 22 (PMP22) and type II single-spanningCitation5 membrane protein myelin protein zero (MPZ) are linked to the major subtypes of demyelinating CMT, type 1A (CMT1A) and type 1B (CMT1B), respectively, which lead to the misfolding of PMP22Citation6-Citation8 and MPZ.Citation9,Citation10 How misfolding of these membrane proteins with different topologies contributes to demyelinating neuropathy remain unknown. We found that misfolded SIMPLE forms abnormal cytosolic aggregates and mediates erroneous interactions with cellular proteins,Citation3 which are pathogenic mechanisms characteristic of many protein-misfolding diseases.Citation11,Citation12 These findings suggest that demyelinating CMT may be a protein-misfolding disease of Schwann cells.
The Proteasome and Aggresome-Autophagy Pathways Protect Schwann Cells Against Toxic Build-Up of Misfolded Proteins
Misfolded myelin proteins, such as PMP22 and MPZ (, step 1a), may be refolded by molecular chaperones at the endoplasmic reticulum (ER)Citation13 (, step 2). When refolding is not possible, these misfolded proteins are retrotranslocated to the cytosol (, step 3) and cleared by the proteasome (, step 4) in a process known as ER-associated degradation (ERAD).Citation14 Our study found that misfolded SIMPLE proteins translocated from the early endosome to the cytosol (, step 1b) are also degraded by the proteasome (, step 4) but through an ERAD-independent mechanism.Citation3 Whether this novel ERAD-independent proteasomal degradation is unique to misfolded SIMPLE or also degrades other C‑tail anchored membrane proteins will need to be addressed by future studies. In addition, the E3 ligases targeting misfolded proteins for ERAD-dependent and -independent clearance of misfolded proteins in Schwann cells remain to be identified.
Figure 1. Protein quality control systems are potential targets for mechanism-based treatments of demyelinating Charcot-Marie-Tooth disease (CMT). Genetic mutations and increased protein expression levels linked to demyelinating CMT induce misfolding of hydrophobic proteins such as peripheral myelin protein 22 (PMP22), myelin protein zero (MPZ), and SIMPLE in Schwann cells (step 1a and 1b). Chaperones at the endoplasmic reticulum (ER) and the cytosol refold misfolded PMP22 and MPZ proteins (step 2), but when refolding is not possible, these proteins are recognized and retrotranslocated to the cytosol by the p97/VCP-dependent ER-associated degradation (ERAD) machinery (step 3). In contrast, misfolded SIMPLE is translocated to the cytosol by an ERAD-independent mechanism (step 1b). These misfolded proteins are targeted to the 26S proteasome by K48-linked poly-ubiquitination for degradation (step 4). When the chaperone and proteasome systems are damaged or overwhelmed, misfolded proteins accumulate and aggregate into toxic oligomers (step 5) that could inhibit proteasome function (step 6) and impair myelination by Schwann cells, leading to demyelinating CMT (step 7). The aggresome-autophagy pathway is another protein quality control system in which misfolded and aggregated proteins are transported by the microtubule-dependent dynein motor complex to the aggresomes (step 8). Aggresomes not only sequester toxic misfolded and aggregated proteins but also recruit autophagic membrane for the formation of autophagosomes (step 9) and subsequent degradation by lysosomal hydrolases upon autophagosome-lysosome fusion (step 10). Therapeutic strategies and currently available agents that enhance the targeting of misfolded and aggregated proteins for turnover are indicated by the color green (upward arrows), and drugs exacerbating the CMT neuropathy phenotype are marked by the color red (downward arrows).
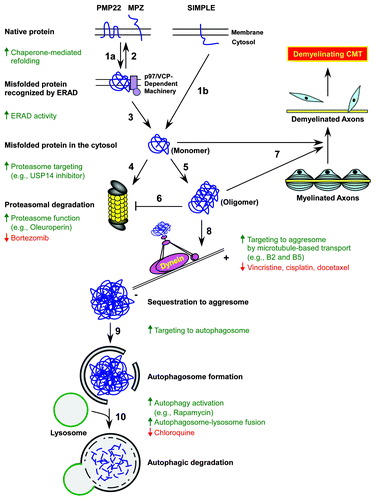
The proteasome system only degrades monomeric misfolded proteins but not aggregated proteins. When proteasome is impaired or overwhelmed, misfolded proteins accumulate and form soluble oligomers (, step 5) that inhibit proteasome function (, step 6) and may impair Schwann cell functions including myelinationCitation13,Citation14 (, step 7). The aggresome-autophagy pathway has emerged as another crucial protein quality control system in Schwann cells that degrades misfolded and aggregated proteins.Citation14-Citation16 Reports by our group and others indicate that Schwann cells handle misfolded PMP22 and SIMPLE by sequestering them into perinuclear aggresomes through a mechanism requiring microtubule-dependent retrograde transportCitation3,Citation14 (, step 8). Although the signaling events that target misfolded PMP22 and SIMPLE into aggresomes have not been identified, our previous findingCitation17 of parkin-mediated K63‑linked poly-ubiquitination in targeting misfolded protein to aggresomes via interactions with the dynein adaptor protein HDAC6 suggest that as yet-to-be identified E3 ligases and adaptor proteins could be responsible for targeting misfolded PMP22 and SIMPLE to aggresomes. Moreover, weCitation3 and othersCitation18 showed that PMP22- and SIMPLE-positive aggresomes are tightly surrounded by autophagosome markers (, step 9) and degraded by autophagy (, step 10).
Impairment in the Proteasome and Aggresome-Autophagy Pathways Contributes to Peripheral Neuropathies
The accumulation of misfolded PMP22 and MPZ at the ERCitation19,Citation20 suggests that ERAD dysfunction and subsequent ER stress may be involved in causing demyelinating CMT. Our recent finding that misfolded SIMPLE is not cleared by ERADCitation3 indicates that CMT pathogenesis can be mediated by dysfunction in ERAD-independent pathways. Proteasome impairment induced by misfolded and aggregated proteins was observed in several mouse models of demyelinating CMT1A.Citation7 Moreover, proteasome inhibition by the chemotherapeutic medication bortezomib causes demyelinating neuropathy and worsens the neuropathic symptoms of CMT patients.Citation21,Citation22 These findings suggest that proteasome dysfunction could contribute to demyelinating CMT pathogenesis.
Impairment of the aggresome-autophagy pathway by microtubule-disrupting drugs such as vincristine, cisplatin, and docetaxel (, step 8) block aggresome-targeting of misfolded PMP22Citation23 and exacerbate the neuropathy phenotype in CMT patients.Citation24 Inhibition of microtubule polymerization observed in Chediak-Higashi syndrome, a genetic disease caused by LYST mutations, leads to severe demyelinating peripheral neuropathy as well.Citation25 Meanwhile, the raise in luminal pH of lysosomes and disruption of autophagosome-lysosome fusion by chloroquineCitation26 also induce peripheral neuropathy.Citation24 These evidence suggests that dysfunction in the aggresome-autophagy pathway is another major contributing factor of demyelinating peripheral neuropathies.
The Proteasome and Aggresome-Autophagy Pathways are Potential Therapeutic Targets in CMT
The studies described above suggest that the augmentation of the proteasome and aggresome-autophagy pathways in Schwann cells could help protect against demyelinating neuropathies. Proteasome activation by oleuroperinCitation27 or enhanced targeting to proteasome by an USP14 inhibitorCitation28 both demonstrated cytoprotective effects. In addition, the chemical compounds B2 and B5 identified from a recent drug screen are inducers of aggresome formation that reduce the cytotoxicity mediated by misfolded proteins.Citation29 These drugs could be evaluated as potential treatments for demyelinating CMT. Our recent finding that autophagy activation by rapamycin promotes autophagic degradation of misfolded SIMPLE,Citation3 together with the reported role of rapamycin in promoting myelination in explant cultures from neuropathic mouse models of CMT1A,Citation30 suggest that autophagy activation could be efficacious for treating demyelinating peripheral neuropathy.
Conclusions
Recent evidence by our group and others has implicated protein misfolding and aggregation in the pathogenesis of demyelinating CMT. The findings that the proteasome and aggresome-autophagy pathways clear misfolded proteins in Schwann cells and their impairment in demyelinating neuropathies suggest that augmentation of these pathways may provide therapeutic benefits for treating demyelinating CMT. Future studies to identify Schwann cell-specific E3 ligases and adaptor proteins that target misfolded proteins for clearance will facilitate the development of new therapeutic strategies that are clinically viable in treating demyelinating CMT.
| Abbreviations: | ||
| CMT | = | Charcot-Marie-Tooth disease |
| CMT1C | = | CMT type 1C |
| PMP22 | = | peripheral myelin protein 22 |
| MPZ | = | myelin protein zero |
| CMT1A | = | CMT type 1A |
| CMT1B | = | CMT type 1B |
| ER | = | endoplasmic reticulum |
| ERAD | = | ER-associated degradation |
Acknowledgments
This work was supported by National Institutes of Health grants NS063501 (SML), NS050650 (LSC), AG034126 (LSC), ES015813 (LL), and GM082828 (LL).
References
- Patzkó A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep 2011; 11:78 - 88; http://dx.doi.org/10.1007/s11910-010-0158-7; PMID: 21080241
- Reilly MM, Murphy SM, Laura M. Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011; 16:1 - 14; http://dx.doi.org/10.1111/j.1529-8027.2011.00324.x; PMID: 21504497
- Lee SM, Olzmann JA, Chin LS, Li L. Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J Cell Sci 2011; 124:3319 - 31; http://dx.doi.org/10.1242/jcs.087114; PMID: 21896645
- Fontanini A, Chies R, Snapp EL, Ferrarini M, Fabrizi GM, Brancolini C. Glycan-independent role of calnexin in the intracellular retention of Charcot-Marie-tooth 1A Gas3/PMP22 mutants. J Biol Chem 2005; 280:2378 - 87; http://dx.doi.org/10.1074/jbc.M405104200; PMID: 15537650
- D'Urso D, Brophy PJ, Staugaitis SM, Gillespie CS, Frey AB, Stempak JG, et al. Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron 1990; 4:449 - 60; http://dx.doi.org/10.1016/0896-6273(90)90057-M; PMID: 1690568
- Fortun J, Go JC, Li J, Amici SA, Dunn WA Jr., Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis 2006; 22:153 - 64; http://dx.doi.org/10.1016/j.nbd.2005.10.010; PMID: 16326107
- Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem 2005; 92:1531 - 41; http://dx.doi.org/10.1111/j.1471-4159.2004.02987.x; PMID: 15748170
- Myers JK, Mobley CK, Sanders CR. The peripheral neuropathy-linked Trembler and Trembler-J mutant forms of peripheral myelin protein 22 are folding-destabilized. Biochemistry 2008; 47:10620 - 9; http://dx.doi.org/10.1021/bi801157p; PMID: 18795802
- Shames I, Fraser A, Colby J, Orfali W, Snipes GJ. Phenotypic differences between peripheral myelin protein-22 (PMP22) and myelin protein zero (P0) mutations associated with Charcot-Marie-Tooth-related diseases. J Neuropathol Exp Neurol 2003; 62:751 - 64; PMID: 12901701
- Mandich P, Fossa P, Capponi S, Geroldi A, Acquaviva M, Gulli R, et al. Clinical features and molecular modelling of novel MPZ mutations in demyelinating and axonal neuropathies. Eur J Hum Genet 2009; 17:1129 - 34; http://dx.doi.org/10.1038/ejhg.2009.37; PMID: 19293842
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 2004; 6:1054 - 61; http://dx.doi.org/10.1038/ncb1104-1054; PMID: 15516999
- Ovádi J, Orosz F, Hollan S. Functional aspects of cellular microcompartmentation in the development of neurodegeneration: mutation induced aberrant protein-protein associations. Mol Cell Biochem 2004; 256-257:83 - 93; http://dx.doi.org/10.1023/B:MCBI.0000009860.86969.72; PMID: 14977172
- Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, et al. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci 1997; 17:7754 - 62; PMID: 9315897
- Ryan MC, Shooter EM, Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis 2002; 10:109 - 18; http://dx.doi.org/10.1006/nbdi.2002.0500; PMID: 12127149
- Olzmann JA, Li L, Chin LS. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem 2008; 15:47 - 60; http://dx.doi.org/10.2174/092986708783330692; PMID: 18220762
- Fortun J, Dunn WA Jr., Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci 2003; 23:10672 - 80; PMID: 14627652
- Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, et al. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol 2007; 178:1025 - 38; http://dx.doi.org/10.1083/jcb.200611128; PMID: 17846173
- Fortun J, Verrier JD, Go JC, Madorsky I, Dunn WA, Notterpek L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol Dis 2007; 25:252 - 65; http://dx.doi.org/10.1016/j.nbd.2006.09.018; PMID: 17174099
- Kamholz J, Awatramani R, Menichella D, Jiang H, Xu W, Shy M. Regulation of myelin-specific gene expression. Relevance to CMT1. Ann N Y Acad Sci 1999; 883:91 - 108; http://dx.doi.org/10.1111/j.1749-6632.1999.tb08572.x; PMID: 10586235
- Lee YC, Lin KP, Chang MH, Liao YC, Tsai CP, Liao KK, et al. Cellular characterization of MPZ mutations presenting with diverse clinical phenotypes. J Neurol 2010; 257:1661 - 8; http://dx.doi.org/10.1007/s00415-010-5590-8; PMID: 20461396
- Hamilton AL, Eder JP, Pavlick AC, Clark JW, Liebes L, Garcia-Carbonero R, et al. Proteasome inhibition with bortezomib (PS-341): a phase I study with pharmacodynamic end points using a day 1 and day 4 schedule in a 14-day cycle. J Clin Oncol 2005; 23:6107 - 16; http://dx.doi.org/10.1200/JCO.2005.01.136; PMID: 16135477
- Filosto M, Rossi G, Pelizzari AM, Buzio S, Tentorio M, Broglio L, et al. A high-dose bortezomib neuropathy with sensory ataxia and myelin involvement. J Neurol Sci 2007; 263:40 - 3; http://dx.doi.org/10.1016/j.jns.2007.05.023; PMID: 17612569
- Watanabe T, Nagase K, Chosa M, Tobinai K. Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br J Cancer 2010; 103:1580 - 7; http://dx.doi.org/10.1038/sj.bjc.6605954; PMID: 20959823
- Weimer LH, Podwall D. Medication-induced exacerbation of neuropathy in Charcot Marie Tooth disease. J Neurol Sci 2006; 242:47 - 54; http://dx.doi.org/10.1016/j.jns.2005.11.014; PMID: 16386273
- Lockman LA, Kennedy WR, White JG. The Chediak-Higashi syndrome: electrophysiological and electron microscopic observations on the peripheral neuropathy. J Pediatr 1967; 70:942 - 51; http://dx.doi.org/10.1016/S0022-3476(67)80267-1; PMID: 4290695
- Spowart J, Lum JJ. Opening a new DOR to autophagy. EMBO Rep 2010; 11:4 - 5; http://dx.doi.org/10.1038/embor.2009.265; PMID: 20033084
- Rigacci S, Guidotti V, Bucciantini M, Parri M, Nediani C, Cerbai E, et al. Oleuropein aglycon prevents cytotoxic amyloid aggregation of human amylin. J Nutr Biochem 2010; 21:726 - 35; http://dx.doi.org/10.1016/j.jnutbio.2009.04.010; PMID: 19616928
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010; 467:179 - 84; http://dx.doi.org/10.1038/nature09299; PMID: 20829789
- Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc Natl Acad Sci USA 2006; 103:4246 - 51; http://dx.doi.org/10.1073/pnas.0511256103; PMID: 16537516
- Rangaraju S, Verrier JD, Madorsky I, Nicks J, Dunn WA Jr., Notterpek L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci 2010; 30:11388 - 97; http://dx.doi.org/10.1523/JNEUROSCI.1356-10.2010; PMID: 20739560