Abstract
Low-voltage-activated T-type calcium channels act as a major pathway for calcium entry near the resting membrane potential in a wide range of neuronal cell types. Several reports have uncovered an unrecognized feature of T-type channels in the control of vesicular neurotransmitter and hormone release, a process so far thought to be mediated exclusively by high-voltage-activated calcium channels. However, the underlying molecular mechanisms linking T-type calcium channels to vesicular exocytosis have remained enigmatic. In a recent study, we have reported that Cav3.2 T-type channel forms a signaling complex with the neuronal Q-SNARE syntaxin-1A and SNAP-25. This interaction that relies on specific Cav3.2 molecular determinants, not only modulates T-type channel activity, but was also found essential to support low-threshold exocytosis upon Cav3.2 channel expression in MPC 9/3L-AH chromaffin cells. Overall, we have indentified an unrecognized regulation pathway of T-type calcium channels by SNARE proteins, and proposed the first molecular mechanism by which T-type channels could mediate low-threshold exocytosis.
Depolarization-evoked synaptic transmission relies on the calcium (Ca2+)-regulated release of quantal packets of neurotransmitters following fusion of synaptic vesicles with the presynaptic plasma membrane.Citation1 It is well established that neuronal voltage-gated Ca2+ channels, by converting electrical signals into intracellular Ca2+ concentration elevations, play a key role in triggering evoked neurotransmitter release.Citation2-Citation4 Hence, Ca2+ entry through high-voltage-activated (HVA) channels (N-, P/Q-, and in some extent and particular cell populations, L- and R-type) into presynaptic nerve terminals in response to action potentials supports a transient Ca2+ microdomainCitation5 essential for synaptic exocytosis. However, the observation that some neurons can release functionally significant amounts of neurotransmitter below the threshold of action potentialsCitation6 questioned the possible involvement of another source of Ca2+ ions, independent of HVA channels activation.
In contrast to HVA channels, low-voltage-activated (LVA) T-type Ca2+ channels activate in response to subthreshold membrane depolarizations between -65 mV and -50 mV and thus represent an important source of Ca2+ entry near the resting membrane potential. Hence, besides controlling important physiological processes by regulating neuronal excitability, pacemaker activity and post-inhibitory rebound burst firing, mounting evidences from various neuronal cell types suggest an efficient role of T-type channels in fast and low-threshold exocytosis.Citation7-Citation10 Until now, however, the mechanism whereby these channels support exocytosis events at the molecular level remained a mystery. In a recent study, we provided compelling evidence of the existence of a Cav3.2/syntaxin-1A molecular complex essential for T-type-dependent exocytosis.Citation11
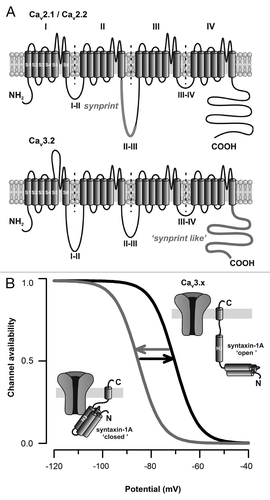
In mammalian synapses, interaction of several members of the vesicle-docking / release machinery (including syntaxin-1A/1B and SNAP-25) onto a synprint (synaptic protein interaction site) domain located within the intracellular loop between domains II and III of the Cav2.1Citation12 and Cav2.2Citation13 channels (, top panel) ensures a close localization of the secretory vesicles near the Ca2+ source. In turn, both synatxin-1A/1B and SNAP-25 modulate calcium channel activityCitation14,Citation15 to fine tune Ca2+ entry and synaptic strength. However, in contrast to HVA channels, all of the three T-type channel members (i.e Cav3.1, Cav3.2 and Cav3.3) lack the consensus synprint site, making the molecular understanding of the involvement of these channels in the exocytosis process quite difficult. Our observation that Cav3.2 channels associate with syntaxin-1A in central neurons prompted us to investigate the possible existence of specific Cav3.2 channel molecular determinants other than the consensus HVA synprint domain. Using biochemical and cellular trafficking approaches, we demonstrated that syntaxin-1A, as well as SNAP-25, interact with the C-terminal domain of Cav3.2 channel (, lower panel). Moreover, using patch-clamp recordings performed on tsA-201 cells expressing Cav3.2 channels, we demonstrated that co-expression of a syntaxin-1A in its “closed” conformational state (i.e the conformation adopted by the syntaxin-1A in isolation or in interaction with Munc18)Citation16,Citation17 potently decreases Cav3.2 channel availability by shifting the voltage-dependence of inactivation toward more hyperpolarized membrane potentials, similarly to what was previously reported for N- and P/Q-type channels.Citation14,Citation15,Citation18-Citation20 Interestingly, this regulation was abolished upon co-expression of SNAP-25, and not observed with a constitutively “open” syntaxin-1A (i.e the conformation adopted upon its association with SNAP-25)Citation18 (). Given that syntaxin-1A undergoes a conformational switch from a “closed” to an “open” conformation during the vesicle release cycle,Citation16,Citation21,Citation22 this suggests that syntaxin-1A may be able to dynamically regulate T-type channel availability during various stages of exocytosis. Interestingly, although T-type channels utilize distinct molecular determinants to interact with SNARE proteins (the C-terminal domain vs. the classical synprint of the II-III linker), they are subjected to a similar SNARE regulation. Does this observation question the molecular mechanism by which binding of syntaxin-1A produces changes in channel gating? Earlier reports have shown that reorganization of intramolecular interactions among the main intracellular loops of Cav2 channels critically influence channel inactivation.Citation23-Citation30 Mapping the intramolecular interactions of T-type channels along with the characterization of the minimal sequence engaged in the interaction with SNARE proteins will provide important structural information on how syntaxin-1A modulates channel gating.
Figure 1. SNARE proteins modulate high- and low-voltage-gated calcium channels via distinct molecular determinants. (A) Membrane topology of voltage-gated calcium channels highlighting the localization of the synprint site located within the intracellular linker between domains II and III of Cav2.1/Cav2.2 channels (top panel), and the “synprint like” domain of Cav3.x channels (bottom panel) located within the C-terminal domain of the channel. (B) Voltage-dependence of Cav3.x channel availability during the conformational switch of syntaxin-1A.
It is well known that direct interaction of SNARE proteins with Cav2.1 and Cav2.2 channels is critical for depolarization-evoked neurotransmitter release. Hence, disruption of the Ca2+ channel-SNARE proteins coupling by deletion of the synprint domain or by peptides derived from the synprint sequence, alters synaptic transmission.Citation31-Citation34 We revealed that similarly to HVA channels, T-type channels-mediated exocytosis relies on a channel-SNARE protein interaction. Indeed, membrane capacitance recordings performed on MPC 9/3L-HA chromaffin cells expressing Cav3.2 channels revealed robust voltage-dependent exocytosis which was totally prevented by co-expression of the Cav3.2 C-terminal domain (i.e the synaptic protein interaction site of Cav3.2). Ablation of Cav3.2-dependent exocytosis most likely results from the specific uncoupling of the channel with SNARE proteins and not from a side alteration of the exocytosis machinery by itself because no alteration was observed when exocytosis was induced by direct intracellular Ca2+ elevation. Hence, we showed that similarly to HVA channels, a physical coupling between SNARE proteins and T-type channels is critical for T-type-dependent exocytosis. Considering the relative small conductance of T-type channelsCitation35 and the restricted diffusion of Ca2+ due to the high Ca2+ buffering capacity of neuronal cells,Citation36 it is conceivable that this interaction allows the close localization of the vesicle-docking / release machinery in close proximity to the Ca2+ source in order to efficiently sense Ca2+ elevation. However, we cannot exclude the possibility that interaction of T-type channels with SNARE proteins could form a macromolecular complex through which channel conformational changes following membrane depolarization would work as an on/off molecular switch of secretion by controlling the ultimate conformational change of the releasing complex as previously proposed for HVA channels.Citation37,Citation38 Although this concept still requires further investigation, the use on a non-conducting channel to investigate T-type-dependent secretion would definitively provide interesting information about the functional importance of T-type channel interaction with SNARE proteins.
Overall, we revealed an unrecognized regulation of low-voltage-activated T-type Ca2+ channels by SNARE proteins, and provide the first evidence for a molecular mechanism by which these channels could mediate low-threshold exocytosis. We revealed that, although T-type Ca2+ channels differ from HVA channels by their molecular constituents, they possess the same ability to functionally interact with SNARE proteins, highlighting a key evolutionary mechanism for specialized fast and spatially delimited exocytosis.
Acknowledgments
N.W is supported by a postdoctoral fellowship from Alberta Innovates Health Solutions (AIHS) and Hotchkiss Brain Institute. G.W.Z is funded by the Canadian Institutes of Health Research, is a Canada Research Chair and AIHS Scientist.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Edwards RH. The neurotransmitter cycle and quantal size. Neuron 2007; 55:835 - 58; http://dx.doi.org/10.1016/j.neuron.2007.09.001; PMID: 17880890
- Llinás R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science 1992; 256:677 - 9; http://dx.doi.org/10.1126/science.1350109; PMID: 1350109
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 2008; 59:861 - 72; http://dx.doi.org/10.1016/j.neuron.2008.08.019; PMID: 18817727
- Weber AM, Wong FK, Tufford AR, Schlichter LC, Matveev V, Stanley EF. N-type Ca2+ channels carry the largest current: implications for nanodomains and transmitter release. Nat Neurosci 2010; 13:1348 - 50; http://dx.doi.org/10.1038/nn.2657; PMID: 20953196
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol 2005; 15:266 - 74; http://dx.doi.org/10.1016/j.conb.2005.05.006; PMID: 15919191
- Ivanov AI, Calabrese RL. Intracellular Ca2+ dynamics during spontaneous and evoked activity of leech heart interneurons: low-threshold Ca currents and graded synaptic transmission. J Neurosci 2000; 20:4930 - 43; PMID: 10864951
- Carabelli V, Marcantoni A, Comunanza V, de Luca A, Díaz J, Borges R, et al. Chronic hypoxia up-regulates alpha1H T-type channels and low-threshold catecholamine secretion in rat chromaffin cells. J Physiol 2007; 584:149 - 65; http://dx.doi.org/10.1113/jphysiol.2007.132274; PMID: 17690152
- Pan ZH, Hu HJ, Perring P, Andrade R. T-type Ca(2+) channels mediate neurotransmitter release in retinal bipolar cells. Neuron 2001; 32:89 - 98; http://dx.doi.org/10.1016/S0896-6273(01)00454-8; PMID: 11604141
- Egger V, Svoboda K, Mainen ZF. Mechanisms of lateral inhibition in the olfactory bulb: efficiency and modulation of spike-evoked calcium influx into granule cells. J Neurosci 2003; 23:7551 - 8; PMID: 12930793
- Tang AH, Karson MA, Nagode DA, McIntosh JM, Uebele VN, Renger JJ, et al. Nerve terminal nicotinic acetylcholine receptors initiate quantal GABA release from perisomatic interneurons by activating axonal T-type (Cav3) Ca²⁺ channels and Ca²⁺ release from stores. J Neurosci 2011; 31:13546 - 61; http://dx.doi.org/10.1523/JNEUROSCI.2781-11.2011; PMID: 21940446
- Weiss N, Hameed S, Fernández-Fernández JM, Fablet K, Karmazinova M, Poillot C, et al. A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem 2012; 287:2810 - 8; http://dx.doi.org/10.1074/jbc.M111.290882; PMID: 22130660
- Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci U S A 1996; 93:7363 - 8; http://dx.doi.org/10.1073/pnas.93.14.7363; PMID: 8692999
- Sheng ZH, Rettig J, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron 1994; 13:1303 - 13; http://dx.doi.org/10.1016/0896-6273(94)90417-0; PMID: 7993624
- Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature 1995; 378:623 - 6; http://dx.doi.org/10.1038/378623a0; PMID: 8524397
- Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci 1999; 2:939 - 41; http://dx.doi.org/10.1038/14721; PMID: 10526329
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, et al. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 1999; 18:4372 - 82; http://dx.doi.org/10.1093/emboj/18.16.4372; PMID: 10449403
- Brunger AT. Structure of proteins involved in synaptic vesicle fusion in neurons. Annu Rev Biophys Biomol Struct 2001; 30:157 - 71; http://dx.doi.org/10.1146/annurev.biophys.30.1.157; PMID: 11340056
- Jarvis SE, Barr W, Feng ZP, Hamid J, Zamponi GW. Molecular determinants of syntaxin 1 modulation of N-type calcium channels. J Biol Chem 2002; 277:44399 - 407; http://dx.doi.org/10.1074/jbc.M206902200; PMID: 12221094
- Wiser O, Bennett MK, Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J 1996; 15:4100 - 10; PMID: 8861939
- Sutton KG, McRory JE, Guthrie H, Murphy TH, Snutch TP. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature 1999; 401:800 - 4; http://dx.doi.org/10.1038/44586; PMID: 10548106
- Fiebig KM, Rice LM, Pollock E, Brunger AT. Folding intermediates of SNARE complex assembly. Nat Struct Biol 1999; 6:117 - 23; http://dx.doi.org/10.1038/5803; PMID: 10048921
- Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 2001; 412:338 - 41; http://dx.doi.org/10.1038/35085583; PMID: 11460165
- Restituito S, Cens T, Barrere C, Geib S, Galas S, De Waard M, et al. The [beta]2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J Neurosci 2000; 20:9046 - 52; PMID: 11124981
- Geib S, Sandoz G, Cornet V, Mabrouk K, Fund-Saunier O, Bichet D, et al. The interaction between the I-II loop and the III-IV loop of Cav2.1 contributes to voltage-dependent inactivation in a beta -dependent manner. J Biol Chem 2002; 277:10003 - 13; http://dx.doi.org/10.1074/jbc.M106231200; PMID: 11790766
- Sandoz G, Lopez-Gonzalez I, Stamboulian S, Weiss N, Arnoult C, De Waard M. Repositioning of charged I-II loop amino acid residues within the electric field by beta subunit as a novel working hypothesis for the control of fast P/Q calcium channel inactivation. Eur J Neurosci 2004; 19:1759 - 72; http://dx.doi.org/10.1111/j.1460-9568.2004.03216.x; PMID: 15078550
- Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron 2005; 46:891 - 904; http://dx.doi.org/10.1016/j.neuron.2005.05.011; PMID: 15953418
- Raghib A, Bertaso F, Davies A, Page KM, Meir A, Bogdanov Y, et al. Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructs. J Neurosci 2001; 21:8495 - 504; PMID: 11606638
- Page KM, Heblich F, Davies A, Butcher AJ, Leroy J, Bertaso F, et al. Dominant-negative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J Neurosci 2004; 24:5400 - 9; http://dx.doi.org/10.1523/JNEUROSCI.0553-04.2004; PMID: 15190113
- Page KM, Heblich F, Margas W, Pratt WS, Nieto-Rostro M, Chaggar K, et al. N terminus is key to the dominant negative suppression of Ca(V)2 calcium channels: implications for episodic ataxia type 2. J Biol Chem 2010; 285:835 - 44; http://dx.doi.org/10.1074/jbc.M109.065045; PMID: 19903821
- Bucci G, Mochida S, Stephens GJ. Inhibition of synaptic transmission and G protein modulation by synthetic CaV2.2 Ca²⁺ channel peptides. J Physiol 2011; 589:3085 - 101; http://dx.doi.org/10.1113/jphysiol.2010.204735; PMID: 21521766
- Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron 1996; 17:781 - 8; http://dx.doi.org/10.1016/S0896-6273(00)80209-3; PMID: 8893034
- Rettig J, Heinemann C, Ashery U, Sheng ZH, Yokoyama CT, Catterall WA, et al. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+ channel/syntaxin interaction. J Neurosci 1997; 17:6647 - 56; PMID: 9254677
- Harkins AB, Cahill AL, Powers JF, Tischler AS, Fox AP. Deletion of the synaptic protein interaction site of the N-type (CaV2.2) calcium channel inhibits secretion in mouse pheochromocytoma cells. Proc Natl Acad Sci U S A 2004; 101:15219 - 24; http://dx.doi.org/10.1073/pnas.0401001101; PMID: 15471993
- Keith RK, Poage RE, Yokoyama CT, Catterall WA, Meriney SD. Bidirectional modulation of transmitter release by calcium channel/syntaxin interactions in vivo. J Neurosci 2007; 27:265 - 9; http://dx.doi.org/10.1523/JNEUROSCI.4213-06.2007; PMID: 17215385
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 2003; 83:117 - 61; PMID: 12506128
- Foehring RC, Zhang XF, Lee JC, Callaway JC. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J Neurophysiol 2009; 102:2326 - 33; http://dx.doi.org/10.1152/jn.00038.2009; PMID: 19675297
- Atlas D. Signaling role of the voltage-gated calcium channel as the molecular on/off-switch of secretion. Cell Signal 2010; 22:1597 - 603; http://dx.doi.org/10.1016/j.cellsig.2010.04.003; PMID: 20388539
- Weiss N. Control of depolarization-evoked presynaptic neurotransmitter release by Cav2.1 calcium channel: old story, new insights. Channels (Austin) 2010; 4:431 - 3; http://dx.doi.org/10.4161/chan.4.6.13613; PMID: 20935476