Abstract
Histone deacetylases (HDACs) are enzymes that catalyse the removal of acetyl groups from a range of nuclear and cytoplasmic proteins. Recently, we described a novel route to neurotrophin-dependent gene activation in neurons, which requires the S-nitrosylation of nuclear HDAC2 by the gaseous molecule nitric oxide (NO) (Nott et al, 2008). We have further investigated the NO-dependent regulation of HDACs in neurons. Using a fluorogenic deacetylation assay, we show that NO decreases the enzymatic activity of a sub-group of neuronal HDACs in vitro and that this inhibition is not due to damaging modifications such as oxidation or tyrosine nitration. The neuronal HDACs whose catalytic activity is inhibited by NO are entirely those that are localised in the cytoplasm. These observations support and extend the concept that nitric oxide is a key regulator of HDAC function in mammalian neurons.
Amongst the genes residing within modern eukaryotic genomes, some are truly ancient, with their origins stretching back to the earliest days of cellular evolution. These genes play fundamental roles in cell biology across the eukaryotic phylogenetic tree. Histone deacetylases are one such family of archaic enzymatic proteins.Citation3 HDACs are found in all modern eukaryotic organisms and are essential for cellular proliferation, differentiation and homeostasis.Citation4 The HDACs are also implicated in the onset and progression of many human diseases which affect an increasingly aging population, such as neurodegenerative disease,Citation5–Citation7 cancer,Citation8,Citation9 cardiovascular diseaseCitation10 and chronic obstructive pulmonary disease.Citation11 The development of effective treatments based on HDAC inhibition is however hampered by a lack of specificity in currently available molecules which often block many, if not all, HDAC isoforms.
We recently described that HDAC2, a nuclear histone deacetylase that is highly expressed in neurons, is regulated by S-nitrosylation.Citation1 NO generated in response to neurotrophin stimulation diffuses throughout the neuronal soma into the cell nucleus and nitrosylates HDAC2 at cysteine residues 262 and 274, releasing it from chromatin to facilitate gene expression.Citation1,Citation2 S-nitrosylation of HDAC2 stimulates its release from DNA but does not alter the enzyme's catalytic activity.Citation1
Histone deacetylases would be more aptly referred to as “lysine deacetylases” as they deacetylate these residues in a wide range of nonhistone proteins, including cytoskeletal proteins and transcription factors.Citation12–Citation14 Mammalian HDACs fall into one of four different classes based on their amino acid sequence and structure.Citation4,Citation15 Class I HDACs are trichostatin A (TSA)-sensitive enzymes, which are typically expressed ubiquitously in mammalian cells and are predominantly nuclear in localization. Class II and IV HDACs are tissue-specific, TSA-sensitive enzymes that may be found in both the nucleus and the cytoplasm where they undergo stimulus-dependent nucleo-cytoplasmic shuttling. Class III HDACs are the sirtuins family. Sirtuins have no homology to Class I/II/IV HDACs and are NAD+, rather than zinc, dependent deacetylases that can be found in the nucleus, cytoplasm or mitochondria.Citation16 The sirtuins are insensitive to inhibitors of class I/II/IV HDACs, such as TSA. HDACs may therefore act in the cytoplasm or nucleus on a range of histone or non-histone proteins to exert their effect on numerous intracellular events.
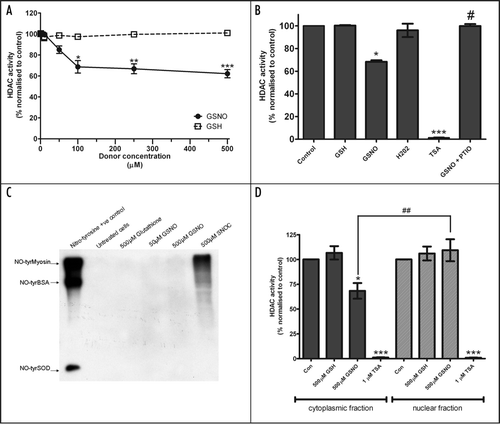
The diversity in HDAC localization and function, in addition to the nature of NO as an ancient and important neuronal signaling molecule,Citation17,Citation18 prompted us to study whether NO-dependent regulation of HDACs might occur at several levels in neurons. Neuronal whole cell lysates were incubated with the donor S-nitrosoglutathione (GSNO), which yields NO in aqueous solution. GSNO inhibited whole-cell histone deacetylase enzymatic activity in a dose-dependent manner (), although glutathione had no effect. Interestingly, GSNO caused only a partial inhibition of whole cell HDAC activity that reached a plateau between 100 and 500 µM concentration. In contrast, the pan-HDAC inhibitor TSA caused a complete (>99%) inhibition of neuronal HDAC activity (). This observation has two important implications: first, the total cellular HDAC activity measured by this assay is dependent upon the TSA-sensitive class I, II and IV HDACs, with no contribution from the TSA-insensitive sirtuins. Second, the partial inhibition of HDAC activity observed upon treatment with GSNO is due to a sub-group of HDACs that account for approximately 40% of total measurable activity. The inhibition is specific to NO, as the effect of GSNO on HDAC activity is completely abolished by the NO-scavenger PTIO (). Oxidative damage and tyrosine nitration has been shown to reduce the enzymatic activity of HDACs.Citation11,Citation19 The decrease in HDAC activity here is unlikely to be due to these damaging post-translational modifications, as the potent oxidizing agent hydrogen peroxide did not cause a significant decrease in HDAC activity () and specific anti-nitrotyrosine antibodies did not detect any modification when neuronal cell lysates were exposed to GSNO (). These results suggest that the change in HDAC activity may occur via a physiological NO-based effect, such as S-nitrosylation. To study whether NO inhibits cytoplasmic and/or nuclear HDACs, NO donors were incubated with either cytoplasmic or nuclear neuronal extracts and subjected to HDAC activity assay. We observed that NO-dependent inhibition of HDAC activity was entirely confined to the cytoplasmic fraction (). In contrast, both cytoplasmic and nuclear fractions were fully inhibited by TSA. One interesting candidate whose catalytic activity may be specifically inhibited by NO is HDAC6, a cytoplasmic class II HDAC that plays a key role in regulating tubulin deacetylation and protein turnover.Citation20–Citation22 However, we were unable to find any NO-induced inhibition of tubulin acetylation (unpublished observations).
Our observations indicate that NO regulates both nuclear and cytoplasmic neuronal HDACs. This may entail HDAC dissociation from promoter regions in the nucleus and inhibition of HDAC catalytic activity in the cytoplasm. A plausible mechanism is that NO regulates the assembly of HDAC-containing macromolecular complexes in neurons through S-nitrosylation of HDACs or HDAC binding partners. Full HDAC enzymatic activity is critically dependent upon their association with other proteins within large macromolecular complexes.Citation23,Citation24 NO may regulate such protein-protein interactions by promoting assembly or disassembly of complexes incorporating S-nitrosylated proteins.Citation25 In the nucleus for example, NO induces the disassembly of complexes containing S-nitrosylated HDAC2 from the chromatin, resulting in decreased histone deacetylation levels and gene transcription. In the cytoplasm, NO may promote the disassembly of HDAC-containing complexes resulting in an inhibition of HDAC catalytic activity.
Our observations provide evidence that NO is a key regulator of multiple HDAC functions in mammalian neurons. As NO may act on HDACs in the nucleus or the cytoplasm, it has the potential to influence multiple intracellular events in neurons. The discovery of a sub-group of NO-sensitive cytoplasmic HDACs implies that events in the cytoplasm, such as cytoskeleton rearrangement or regulation of protein stability and turnover, could potentially be modulated through NO-dependent changes in HDAC activity. Finally, the identification of NO as a modulator of specific HDACs in neurons may also prove useful for targeting those isoforms that are involved in the progression of neurodegenerative disease.
Figures and Tables
Figure 1 (A) Whole cell lysates prepared from E18 embryonic cortical neurons were used in a fluorogenic HDAC assay as previously shown.Citation1,Citation26 Lysates were incubated with the NO donor GSNO, or control glutathione (GSH) for 1 hour at room temperature before HDAC activity was assayed. HDAC activity was normalised to untreated control (100% activity). Data were analysed using paired t-tests comparing GNSO vs. GSH-treated lysate at each concentration point (n = 5). *p = 0.0161; **p = 0.0085; ***p = 0.0006. (B) Whole cell neuronal lysates were incubated with GSNO, GSH, hydrogen peroxide(all 500 µM), TSA (1 µM) and GSNO (500 µM) in the presence of the NO scavenger PTIO (200 µM) for one hour at room temperature. Data are normalized to untreated (100%) and presented as mean ± SEM (n = 3): *p < 0.05 and ***p < 0.001 (versus control), #p < 0.05 (GSNO vs. GSNO + PTIO) by one-way ANOVA with Tukey's post hoc test. (C) Anti-nitrotyrosine western blot analysis of neuronal lysates following 1 hour incubation with NO donors. Tyrosine-nitrated myosin, BSA and superoxide dismutase were used as positive control. Tyrosine nitration was not observed with concentrations of GSNO up to 500 µM (n = 2). Some tyrosine nitration appeared following treatment with S-nitrosocysteine (SNOC). (D) Neuronal cytoplasmic and nuclear extracts were incubated with GSNO (500 µM), GSH (500 µM) or TSA (1 µM) for 1 hour at room temperature and subjected to fluorogenic HDAC assay. Data are presented as mean ± SEM (n = 3) after normalisation to untreated (100%) HDAC activity. Statistical differences were calculated using one-way ANOVA with Tukey's post hoc test: *p < 0.05 and ***p < 0.001 (versus control), ##p < 0.01 (500 µM GSNO vs. 500 µM GSH).
Acknowledgements
We would like to thank Andrew Porter for assistance in carrying out experiments analyzing the effects of nitric oxide on tubulin acetylation. This work was supported by the Medical Research Council (Research Grant G0500792) and the European Research Council (Marie Curie International Reintegration Grant MIRG-CT-2005-016501). P. Marc D. Watson is the recipient of a Medical Research Council Career Development Fellowship. Antonella Riccio is a recipient of an MRC Career Development Award (G117/533).
Addendum to:
References
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 2008; 455:411 - 415
- Riccio A, Alvania RS, Lonze BE, Ramanan N, Kim T, Huang Y, et al. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell 2006; 21:283 - 294
- Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 2004; 338:17 - 31
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nature Rev 2008; 9:206 - 218
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 2008; 7:854 - 868
- Hahnen E, Hauke J, Trankle C, Eyupoglu IY, Wirth B, Blumcke I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs 2008; 17:169 - 184
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol 2008; 8:57 - 64
- Mottet D, Castronovo V. Histone deacetylases: target enzymes for cancer therapy. Clin Exp Metastasis 2008; 25:183 - 189
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26:5420 - 5432
- Berry JM, Cao DJ, Rothermel BA, Hill JA. Histone deacetylase inhibition in the treatment of heart disease. Expert Opin Drug Saf 2008; 7:53 - 67
- Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Ann Rev Physiol 2008; In press http://dx.doi.org/10.1146/annurev.physiol.010908.163257
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene 2005; 363:15 - 23
- Carey N, La Thangue NB. Histone deacetylase inhibitors: Gathering pace. Curr Op Pharmacol 2006; 6:369 - 375
- Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Molecular cell 2008; 31:449 - 461
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370:737 - 749
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J 2007; 404:1 - 13
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 2007; 8:766 - 775
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci 2008; 27:2783 - 2802
- Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Comm 2004; 315:240 - 245
- Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008; 18:291 - 297
- Sadoul K, Boyault C, Pabion M, Khochbin S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 2008; 90:306 - 312
- Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007; 26:5468 - 5476
- Walkinshaw DR, Tahmasebi S, Bertos NR, Yang XJ. Histone deacetylases as transducers and targets of nuclear signaling. J Cell Biochem 2008; 104:1541 - 1552
- Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem 2004; 93:57 - 67
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature Rev 2005; 6:150 - 166
- Wegener D, Wirsching F, Riester D, Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol 2003; 10:61 - 68