Abstract
Over 130 million people world-wide are chronically infected with hepatitis C virus (HCV). New antiviral treatment strategies are needed due to limitations with current therapy. The identification of cellular cofactors of infection has the potential to broadly expand our therapeutic targets. We recently reported an RNA interference screen of host membrane trafficking genes in HCV infection and replication and identified several cellular co-factors for viral replication. Phosphatidylinositol 4-kinase III alpha (PI4K-IIIα) was found to be essential for HCV replication. PI4K-IIIα co-localized with viral replication markers. Silencing of PI4K-IIIα by siRNAs prior to HCV infection prevented rearrangement of intracellular membranes associated with viral replication complexes, termed the membranous web. Our data suggest that PI4K-IIIα is involved in establishing HCV replication complexes, however the mechanism is unknown. From our analysis, along with several other studies that have identified cellular cofactors for HCV replication, we propose that PI4K-IIIα may nucleate replication complex formation by facilitating the interaction of viral and/or cellular proteins with cellular membrane-associated phospholipids.
Hepatitis C virus (HCV) is a significant human pathogen associated with chronic liver infection, liver cirrhosis and hepatocellular carcinoma. A vaccine is not available and the current therapy of ribavirin and interferon is effective in only half of the treatments.Citation1 This has led to a major push to develop novel antiviral strategies. The vast majority of candidate therapies in clinical trials target viral enzymatic functions, typically either NS5B, the viral RNA-dependent RNA polymerase, or the HCV protease NS3. Like all viruses, HCV relies heavily on the host cell to replicate. As such, there is significant interest in trying to identify cellular genes that are required for HCV infection, both to understand the basic biology of the HCV life cycle and to unearth potential new therapeutic targets.
A number of groups have published RNA interference (RNAi) screens designed to identify cellular cofactors required for HCV infection.Citation2–Citation7 As with similar approaches investigating HIV cofactors,Citation8–Citation10 there has been a relatively small overlap of genes identified in these studies. There are numerous reasons for the varied results, including differences in RNAi libraries, cells, transfection protocols, HCV genotypes and HCV replication systems (sub-genomic replicons versus infectious HCV). In our hands, all of these differences can have a pronounced impact on the penetrance of an RNAi phenotype. In particular, we find that the infectious HCV system produces more robust phenotypes than replicons in our RNAi studies. Another source of variability that arises between lists of identified HCV cofactors is the cut-off for significance. In most genome-wide RNAi screens, the RNAi phenotype needs to be quite large to register as significant (>2 standard deviations). Since RNAi produces only partial phenotypes, many genes that are relevant to HCV infection can be overlooked due to incomplete inhibition by RNAi. Therefore, we prefer to view lists of genes from different studies as inclusive, as opposed to limiting the lists to common denominators through strict meta-analysis. In the end, the value of cofactor lists is to serve as a resource for generating hypotheses that can be tested further by multiple lines of experimentation.
Despite our stated preference against meta-analysis of RNAi results, we and at least two other groups have identified a common cellular cofactor of HCV infection: phosphatidylinositol 4-kinase III α (PI4K-IIIα).Citation5–Citation7 Encoded by the PIK4CA gene and localized primarily to the endoplasmic reticulum (ER), PI4K-IIIα is a lipid kinase whose main cellular function is to generate phosphatidylinositol 4-monophosphate, or PI(4)P. Following our initial characterization of the function of PI4K-IIIα in HCV replication, we proposed that it plays a role in establishing HCV replication complexes, which consist of the replication machinery married to unique membranous structures induced by HCV infection. Membrane rearrangements are hallmarks of all positive-stranded RNA virus infections.Citation11,Citation12 In the case of HCV infection, these structures are clusters of intracellular, cytosolic membranes and are a major pathology associated with replication of the viral RNA genome in vivo.Citation13,Citation14 Similar membranous structures are observed using in vitro HCV replication and viral protein expression systems and have been termed “membranous webs”.Citation14–Citation16 Evidence that HCV replication proteins and de novo synthesized viral RNA localize to the membranous web,Citation14,Citation15 which forms in close proximity to the ER, implicates the webs as sites of viral replication and the ER as a potential membrane source. It is unclear whether membrane rearrangements serve primarily to provide a high local concentration of replication components, provide a scaffold for replication, or shield the viral RNAs from recognition by the innate immune system. The mechanism of HCV replication complex formation, including intracellular membrane rearrangements, remains to be determined.
Our proposal that PI4K-IIIα functions in HCV replication complex formation was based on the following data. Treatment with siRNAs and pharmacological inhibition of PI4K-IIIα reduces HCV replication. PI4K-IIIα is not required for HCV entry or initial translation of the viral genomic RNA. It co-localizes with markers of the HCV replicase, and most importantly, membranous webs fail to accumulate in cells that have been silenced for PI4K-IIIα expression. A role for PI4K-IIIα in forming replication complexes is also supported by elegant studies from Tai et al. who show that the HCV replicase-associated NS5A protein has aberrant localization in cells that have inducible expression of the full HCV polyprotein and have been silenced for PI4K-IIIα expression.Citation6 A key future experiment is to test whether PI4K-IIIα is required for membranous web formation by expression of HCV NS4B, the replicase protein sufficient for inducing these membrane alterations.
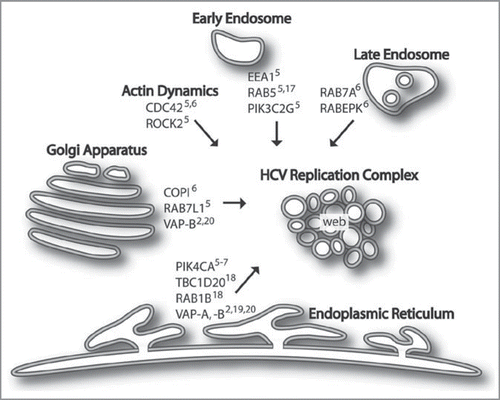
The main question remaining is the mechanism of HCV replication complex formation. RNAi analysis has identified a number of cellular candidates that may be involved in constructing membrane-associated sites of replication (see ). These include early endosomes (EEA1,Citation5 RAB5Citation5,Citation17 and PIK3C2GCitation5), late endosomes (RAB7A,Citation6 RABEPKCitation6), vesicles associated with the Golgi apparatus (COPI componentsCitation6 and RAB7L1Citation5), and the ER (PIK4CA,Citation5–Citation7 TBC1D20Citation18 and RAB1BCitation18). Components of vesicular trafficking and membrane fusion (VAP-ACitation2,Citation19 and VAPBCitation2,Citation20) and regulators of actin reorganization (CDC42Citation5,Citation6 and ROCK2Citation5) may also be involved. A role in replication complex formation for EEA1, RAB5, RAB7, COPI subunits, PIK4CA, TBC1D20, VAP-A, VAP-B and CDC42 is further supported by microscopy, proteomic and/or protein biochemistry studies.Citation5,Citation17,Citation19–Citation24 These cofactors, in addition to others, may work in combination to establish the membrane-associated replication complexes in infected cells.
One hypothesis is that phosphorylation of phosphatidylinositol (PI) molecules by PI4K-IIIα and subsequent downstream modifications of PI(4)P attract cellular and/or viral proteins to phospholipids. This serves to nucleate replication proteins and membrane-bound vesicles, potentially establishing the membranous web, which appears to be a non-uniform, heterogeneous mix of vesicles. Additionally, PI4K-IIIα itself may be directly required for establishing and maintaining an intimate interaction of the HCV replicase with cellular membranes. This is suggested from yeast two-hybrid analysis wherein PI4K-IIIα interacted with HCV NS5A.Citation22
In addition to our interest in the biology of PI4K-IIIα in HCV infection, it is possible that PI4K-IIIα may be a legitimate drug target for treating HCV infection. Pharmacological inhibitors of PI kinase activity prevent HCV replication in vitroCitation5,Citation6 and PI-3 kinase inhibitors have been successful therapies against certain cancers.Citation25 We speculate that inhibitors specific to PI4K-IIIα may be successful therapeutics for HCV with fewer issues of resistance than is observed for drugs targeting viral enzymes.
Figures and Tables
Figure 1 Host factors and membrane compartments proposed to be involved in membrane-associated HCV replication complex formation. Several RNAi-validated cellular cofactors for HCV replication have been identified as shown. HCV infection leads to membrane rearrangements, forming a structure termed the membranous web thought to be the site of viral replication. We hypothesize that proteins and membranes from endosomes, Golgi and ER contribute to replication complex formation. Particularly, PI4K-IIIα, which is encoded by the PIK4CA gene, is critically important for HCV replication. Its function may be required to nucleate viral or cellular proteins and vesicles to establish the membrane-associated sites of viral replication.
Addendum to:
References
- Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975 - 982
- Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA 2007; 104:12884 - 12889
- Ng TI, Mo H, Pilot-Matias T, He Y, Koev G, Krishnan P, et al. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology 2007; 45:1413 - 1421
- Supekova L, Supek F, Lee J, Chen S, Gray N, Pezacki JP, et al. Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J Biol Chem 2008; 283:29 - 36
- Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA 2009; 106:7577 - 7582
- Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009; 5:298 - 307
- Vaillancourt FH, Pilote L, Cartier M, Lippens J, Liuzzi M, Bethell RC, et al. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009; 387:5 - 10
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008; 319:921 - 926
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008; 135:49 - 60
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008; 4:495 - 504
- Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 2008; 6:363 - 374
- Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat Rev Microbiol 2006; 4:371 - 382
- Pfeifer U, Thomssen R, Legler K, Bottcher U, Gerlich W, Weinmann E, et al. Experimental non-A, non-B hepatitis: four types of cytoplasmic alteration in hepatocytes of infected chimpanzees. Virchows Arch B Cell Pathol Incl Mol Pathol 1980; 33:233 - 243
- Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 2002; 76:5974 - 5984
- Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 2003; 77:5487 - 5492
- Rouille Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, et al. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol 2006; 80:2832 - 2841
- Stone M, Jia S, Do Heo W, Meyer T, Konan KV. Participation of Rab5, an early endosome protein, in hepatitis C virus RNA replication machinery. J Virol 2007; 81:4551 - 4563
- Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J Biol Chem 2007; 282:36354 - 36361
- Gao L, Aizaki H, He JW, Lai MM. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 2004; 78:3480 - 3488
- Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, et al. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J Virol 2005; 79:13473 - 13482
- Mannova P, Fang R, Wang H, Deng B, McIntosh MW, Hanash SM, et al. Modification of host lipid raft proteome upon hepatitis C virus replication. Mol Cell Proteomics 2006; 5:2319 - 2325
- Ahn J, Chung KS, Kim DU, Won M, Kim L, Kim KS, et al. Systematic identification of hepatocellular proteins interacting with NS5A of the hepatitis C virus. J Biochem Mol Biol 2004; 37:741 - 748
- Sklan EH, Staschke K, Oakes TM, Elazar M, Winters M, Aroeti B, et al. A Rab-GAP TBC domain protein binds hepatitis C virus NS5A and mediates viral replication. J Virol 2007; 81:11096 - 11105
- Tu H, Gao L, Shi ST, Taylor DR, Yang T, Mircheff AK, et al. Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 1999; 263:30 - 41
- Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin 2009; 59:111 - 137