Abstract
Localization microscopy techniques are super-resolution fluorescence imaging methods based on the detection of individual molecules. Despite the relative simplicity of the microscope setups and the availability of commercial instruments, localization microscopy faces unique challenges. While achieving super-resolution is now routine, issues concerning data analysis and interpretation mean that revealing novel biological insights is not. Here, we outline why data analysis and the design of robust test samples may hold the key to harness the full potential of localization microscopy.
It has been three years since Nature Methods pronounced super-resolution fluorescence microscopy, or nanoscopy, as the Method of the Year in 2008 (Nature Methods, 2009). Since its invention, the technique of localizing individual fluorescent molecules in a densely labeled sample by exploiting light-dark transitions has been extended to live cell,Citation1 multi-colorCitation2-Citation4 and 3D imagingCitation5-Citation7 and manufacturers are now offering user-friendly, fully integrated instruments (e.g., Nikon’s N-STORM, Zeiss’ Elyra PALM, Leica’s SR GSD and Applied Precision’s imminent instrument called Monet). Despite these recent advances, we have only scratched the surface of what localization microscopy could achieve for the biological sciences. To record biological process on the single molecule level will give a whole new meaning to the very concept of molecular biology; potentially allowing us to quantify the number of proteins in time and space and hence assign function to individual molecules.
It seems almost too good to be true that this breakthrough technology is built on conventional fluorescence microscopes equipped with lasers and a sensitive camera– equipment that has already been used for wide-field fluorescence imaging and single-particle tracking experiments for decades. Yet, with the right fluorophores and analysis software, localization microscopy can image biological samples with high molecular densities without compromising the localization accuracy of single fluorophore. So what is stopping localization microscopy from becoming a routine research tool?
Localization microscopy differs from all other fluorescence imaging techniques in that an image is built up literally molecule-by-molecule. This is achieved by the stochastic activation of a sparse subset of the molecules in a field-of-view such that the point-spread-functions (PSFs) obtained when imaging individual fluorophores can be recorded and analyzed. By repeated cycles of activation or switching, localization and bleaching, the coordinates of many single molecules are obtained. This is possible because algorithms can identify the center of the non-overlapping PSFs of individual molecules (). In addition to the localization coordinates, other parameters such as the number of photons emitted per molecule, the width of the PSF and the fluorescent background in the immediate vicinity of the detected molecule are recorded from which fluorophore characteristics such as on-off duty cycles and bleaching rates can be extracted. These parameters can be exceedingly useful in assessing the performance of fluorophores and the quality of the microscope and data, and can in fact contain novel information. Practical guides on how to implement the localization microscopy and select suited experimental conditions have now been published.Citation8-Citation10 The key question is how do we go from detection of individual molecules to new biological insights, and how do we make sure no information is lost or artifacts introduced during the various steps of recording, analysis and interpretation.
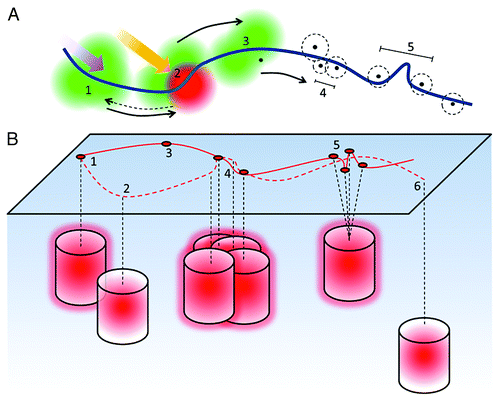
Figure 1. Principles of localization microscopy. (A) Fluorophores in close proximity an underlying structure, here depicted as a fiber (blue), are illuminated with low intensity photo-converting or photo-switching light (1). This causes them to reversibly or irreversibly switch/convert into a sparse array of PSFs, which are recorded and analyzed (2). After localization of the center of the PSFs, molecules are either irreversibly bleached or returned to the initial ‘off-state’ and the process repeated many times (3). This results in a series of x-y coordinates, each with an associated localization precision (dashed line) which determines the optical resolution - the closest separation of resolvable fluorophores (4). However, if the detected spot density is low, this can limit the obtainable structural resolution (5). (B) In an ideal case, one fluorophore delivers one highly localized coordinate (1). Sub-optimal setting of the intensity detection threshold, however, can lead to false negatives (2) or false positives (3). Localizations can also be missed when the cluster density is very high or the fluorophore has a high duty cycle due to overlapping PSFs (4). Additional localizations can appear in a cluster due to single molecule on-off cycling and the associated localization precisions (5). Molecules may also be missed due to their depth in the evanescent field (6). All these effects can lead to misinterpretation (solid line) of the underlying structure (dashed line), shown here as a fiber.
The Acronym Jungle
Often the first question non-experts ask themselves is whether there is a difference between (F)PALM,Citation11,Citation12 PALMIRA,Citation13 (d)STORM,Citation14,Citation15 GSDIM,Citation16 BALMCitation17 and (u)PAINT.Citation18,Citation19 The diversity of acronyms is particularly confusing since the commercial instruments are marketed along these lines. These acronyms represent fundamentally the same technique and are generally used based on the method employed to induce photo-switching or blinking.Citation20 F(PALM) and (d)STORM for example use photo switchable/convertible fluorophores or pairs of fluorophores whereas GSDIM uses ground state depletion by ‘parking’ the fluorophores in a long-lived triplet state to effectively turn fluorophores on and off. BALM and (u)PAINT use the detection of molecules as they become immobilised/activated while binding to a structure. In an attempt to unify these imaging approaches, we refer to them collectively as “localization microscopy” methods based on the localization of molecules in a densely labeled sample by exploiting on-off transitions.
All localization microscopy techniques rely on the temporal separation of fluorescence emission, which is be achieved either by switching between a dark and fluorescent state or by consecutive binding of individual fluorophores to the structure. A probe that switches only once is advantageous when quantifying the absolute number of fluorescent molecules in a sample since each fluorophore is counted only once. Most organic dyes however, can be photo switched reversibly and hence are localized multiple times. In reality, a single fluorescent protein will also appear as a cluster of localizations due to variable intervals of blinking before irreversible photobleaching.Citation21-Citation24 In fact, it should be emphasized that the number of switching cycles is a critical parameter in localization microscopy as it impacts on reproducibility and quantification of super-resolution imaging data. Indeed, multiple blinking of fluorophores can be advantageous (especially in time-resolved imaging) as it allows the same structure to be sampled multiple times.
What is “Resolution”: Optical vs. Structural Resolution
Because localization microscopes are super-resolution microscopes, the first ‘proof’ is generally to evaluate the optical resolution, defined as the shortest distance at which two point emitters can be distinguished (the Rayleigh Criterion). However, the resolution does not only depend on the nature of the probe but also on the labeling density or expression level. For example, if a protein is labeled with an antibody with low affinity or a fusion protein is expressed at a low copy numbers, the structural resolution, i.e., the finest resolvable level of detail in a continuous structure, will be lower because the molecules are too sparsely distributed (). Many papers report the average localization precision, which reflects only the optical quality of the microscope and the nature of the fluorophore. However, localization precision should not be conflated with resolution.
The aim of most biological imaging tools is to extract structural details, molecular distributions and interactions. Contrary to many classical microscopy methods, the structural resolution in localization microscopy is potentially much lower than the optical resolution. In all microscopy, the labeling density must be sufficiently high to comply with the Nyquist-Shannon sampling theorem stating that the mean distance between neighboring molecules (the sampling interval) must be at least half the stated resolution (). In localization microscopy, the optical resolution (localization precision) can be so high that the labeling density becomes the factor limiting resolution. It should be noted that as the optical resolution continues to improve toward around 10 nm and beyond, the physical size of the antibodies or fluorescent proteins may start to become the limiting factor for determining resolution. It is impossible to fully exploit the high localization precision of a small fluorescent probe if it is attached to, for example a bulky antibody. The direct labeling of a protein of interest with organic dyes in heterogeneous samples is an attractive option,Citation10,Citation25,Citation26 but so far, the genetic tags of these strategies are of similar size as fluorescent proteins and many of the dyes themselves are cell impermeable.
Another limiting factor is the imaging speed, which must be faster than the sample dynamics to enable the reconstruction of statistically confirmed super-resolved images.Citation27 Whether or not a dynamic process can be temporally resolved is mainly controlled by two parameters: the complexity of the structure and its labeling density, and the photo-switching rates of the fluorophores. Imaging at higher speeds requires an increase in the number of localizations per frame and area, i.e., the fluorescent spot density. Here, promising new alternatives are on the horizon, such as the DAOSTORM algorithm, which enable multiple kernels to be fitted to overlapping PSFsCitation28 and other multiple-emitter analysis procedures.Citation29 A recently described approach takes multiple-emitter analysis a step further and models wide-field images.Citation30 The method, termed Bayesian localization microscopy, calculates models that fit the most likely fluorophore distribution in the data set considering fluorophore blinking and bleaching processes, and then averages multiple models to represent the underlying structure.
From Seeing to Quantification
The issue of fidelity in localization microscopy has been largely ignored by the scientific community as well as the manufacturers. Since localization microscopy builds up images molecule-by-molecule, we have to ask how many fluorescent molecules are missed or artificially added. The problems of maturation of fluorescent proteins, the fact that an unknown fraction of the proteins are actually photo-switchable and the level of replacement of endogenous protein with tagged proteins all influence the fraction of fluorescent species. But localization microscopy also faces the additional problem that if too many fluorescent molecules are missed because of the low number of photons emitted, inappropriately set switching/activation rates and photo bleaching, the resulting image is incomplete and can be wrong, rather than simply of low quality (). This problem arises because it is generally not known with what efficiency single molecules are activated. Nor is it generally known with what efficiency they are detected over background.
Hence the question remains: can localization microscopy be used for protein quantification and extract reliable numbers of molecules that exist in specific subcellular compartments at a given time? Intuitively, one may expect this is possible with fluorescent proteins, which are expected to be irreversibly bleached after photo-activation. However, fluorescent proteins, like organic dyes also exhibit transitions to long-lasting non-fluorescent off states and can therefore appear in multiple frames.Citation21,Citation23,Citation31 One approach is to take advantage of the time dependence of photo switching by analyzingCitation32 and adjusting the number frames in which no fluorescent molecule appears within an area before events are counted as separate molecules. The blinking can also be factored into the analysis method directly using techniques such as pair correlation.Citation22,Citation24
Other parameters like sensitization of fluorophores in complexes cannot be controlled post acquisition. Hence, strategies and test samples have to be developed that allow us to measure the labeling efficiency and the percentage of detected molecules under the applied experimental conditions. Recently, Dempsey et al. provided a strategy of how to select appropriate fluorescent dyes that yield high x and y localization precisions and structural resolution and are therefore ideal to detect subcellular structures as demonstrated on clathrin-coated pits.Citation10 Samples for protein quantifications are more difficult to engineer. Such samples might comprise of well-defined dimer or multimer structures such as the nuclear pore complex with an 8-fold symmetrical structureCitation33 () or 3D DNA origamiCitation34 carrying a defined number of fluorophores.
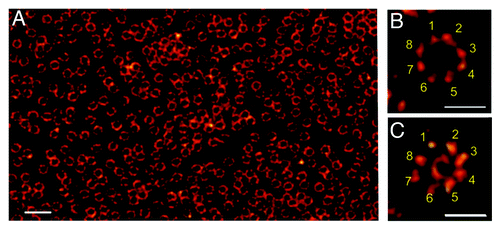
Figure 2. Nuclear envelope of a Xenopus laevis oocyte as seen by dSTORM.Citation33 (A) Nuclear envelopes isolated from Xenopus laevis oocytes were labeled with Alexa647 by indirect immunofluorescence against gp210, a protein that localizes to the lumen of the nuclear envelope bordering the pore wall. (B) Higher magnifications reveal the structural arrangement of gp210 proteins in nuclear pore complexes (NPCs). The 8-fold symmetry of the gp210 ring around each NPC (B) and the diameter of the central channel of ~40 nm is correctly identified with an optical resolution of ~15 nm (C) using WGA-Alexa647 binding to nucleoporins of the central channel (ref. Citation33). Scale bars: 500 nm (A), 150 nm (B, C).
The development of biological test samples would be of great benefit in establishing localization microscopy as a routine instrument. Not only would such samples standardize imaging conditions between laboratories and establish milestones for manufacturers, but they would also highlight the effect of post-acquisition thresholding on the final data set. Recently we showed that in a TIRF set up, thresholding by the number of photons emitted and background, parameters that determine the localization precision, yields a different result than thresholding by the localization precision directly.Citation35 Hence post-acquisition data manipulation can include or exclude a selected subset of molecules, which of course influences the data interpretation. In localization microscopy, the stringency of single molecule detection always influences the structural resolution. If the thresholds are set low so that a high proportion of the fluorescent events are included in the final data set, the probability that some events are background and not genuine molecules is increased. If the thresholds for single molecule detection are high, the structural resolution may not be sufficiently high for a positive identification of the underlying structure.
Data Interpretation
Localization microscopy generates images that we have never seen before. Our eyes are trained to find patterns and we often focus on the biggest and brightest structures in an image. However, localization microscopy images are mathematically generated images. The size and brightness of dots represents user-selected parameters. So how can we represent localization microscope ‘images’ without influencing the viewer? To date, there have been two mathematical approaches to describe the distribution of molecules in a two-dimensional field. Ripley’s K-function analysis compares distributions relative to random distributions and has the advantage that the degree of non-randomness can be assigned to each molecule so that cluster maps can be established.Citation36 An alternative approach is pair-correlation analysis,Citation22 which determines the probability of finding a molecule at a given distance from another molecule compared with the probability expected from random distribution of molecules.
These are only two examples that both quantify molecular distributions. Clearly, we need new algorithms to analyze localization microscopy data sets to identify underlying biological entities including continuous structures and boundaries. But as Bayesian localization microscopyCitation30 demonstrates, novel analysis methods need to be extensively tested against simulations and, ideally, biological test samples. New approaches, however, also afford an opportunity to think differently about biological structures in general, in which inherently dynamic molecular interactions yield stable entities. In fact, we may need to refine, in mathematical terms, what we mean by stable entities, assembly and disassembly so that we develop a new language to describe biological processes on the molecular level. After all, with localization microscopy, the limitation no longer lies in the generation of an ‘image’ but in the extraction of its biological meaning.
Acknowledgments
We acknowledge funding from the Australian Research Council (D.M.O., K.G.), National Health and Medical Research Council of Australia (K.G.), Deutsche Forschungsgesellschaft, Grant SA829/8-1 (M.S.) and Human Frontier Science Program (K.G.).
References
- Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods 2008; 5:417 - 23; http://dx.doi.org/10.1038/nmeth.1202; PMID: 18408726
- Bock H, Geisler C, Wurm CA, von Middendorf C, Jakobs S, Schönle A, et al. Two-color far-field fluorescence nanoscopy based on photoswitchable emitters. Appl Phys B 2007; 88:161 - 5; http://dx.doi.org/10.1007/s00340-007-2729-0
- Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A 2007; 104:20308 - 13; http://dx.doi.org/10.1073/pnas.0710517105; PMID: 18077327
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 2007; 317:1749 - 53; http://dx.doi.org/10.1126/science.1146598; PMID: 17702910
- Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008; 319:810 - 3; http://dx.doi.org/10.1126/science.1153529; PMID: 18174397
- Jones SA, Shim SH, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat Methods 2011; 8:499 - 508; http://dx.doi.org/10.1038/nmeth.1605; PMID: 21552254
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, et al. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat Methods 2008; 5:527 - 9; http://dx.doi.org/10.1038/nmeth.1211; PMID: 18469823
- Gould TJ, Verkhusha VV, Hess ST. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat Protoc 2009; 4:291 - 308; http://dx.doi.org/10.1038/nprot.2008.246; PMID: 19214181
- van de Linde S, Löschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat Protoc 2011; 6:991 - 1009; http://dx.doi.org/10.1038/nprot.2011.336; PMID: 21720313
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods 2011; 8:1027 - 36; http://dx.doi.org/10.1038/nmeth.1768; PMID: 22056676
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006; 313:1642 - 5; http://dx.doi.org/10.1126/science.1127344; PMID: 16902090
- Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 2006; 91:4258 - 72; http://dx.doi.org/10.1529/biophysj.106.091116; PMID: 16980368
- Egner A, Geisler C, von Middendorff C, Bock H, Wenzel D, Medda R, et al. Fluorescence nanoscopy in whole cells by asynchronous localization of photoswitching emitters. Biophys J 2007; 93:3285 - 90; http://dx.doi.org/10.1529/biophysj.107.112201; PMID: 17660318
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 2006; 3:793 - 5; http://dx.doi.org/10.1038/nmeth929; PMID: 16896339
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, et al. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl 2008; 47:6172 - 6; http://dx.doi.org/10.1002/anie.200802376; PMID: 18646237
- Fölling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods 2008; 5:943 - 5; http://dx.doi.org/10.1038/nmeth.1257; PMID: 18794861
- Schoen I, Ries J, Klotzsch E, Ewers H, Vogel V. Binding-activated localization microscopy of DNA structures. Nano Lett 2011; 11:4008 - 11; http://dx.doi.org/10.1021/nl2025954; PMID: 21838238
- Giannone G, Hosy E, Levet F, Constals A, Schulze K, Sobolevsky AI, et al. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys J 2010; 99:1303 - 10; http://dx.doi.org/10.1016/j.bpj.2010.06.005; PMID: 20713016
- Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci U S A 2006; 103:18911 - 6; http://dx.doi.org/10.1073/pnas.0609643104; PMID: 17142314
- Lippincott-Schwartz J, Manley S. Putting super-resolution fluorescence microscopy to work. Nat Methods 2009; 6:21 - 3; http://dx.doi.org/10.1038/nmeth.f.233; PMID: 19116610
- Endesfelder U, Malkusch S, Flottmann B, Mondry J, Liguzinski P, Verveer PJ, et al. Chemically induced photoswitching of fluorescent probes--a general concept for super-resolution microscopy. Molecules 2011; 16:3106 - 18; http://dx.doi.org/10.3390/molecules16043106; PMID: 21490558
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods 2011; 8:969 - 75; http://dx.doi.org/10.1038/nmeth.1704; PMID: 21926998
- Annibale P, Scarselli M, Kodiyan A, Radenovic A. Photoactivatable Fluorescent Protein mEos2 Displays Repeated Photoactivation after a Long-Lived Dark State in the Red Photoconverted Form. Journal of Physical Chemistry Letters 2010; 1:1506 - 10; http://dx.doi.org/10.1021/jz1003523
- Veatch SL, Machta BB, Shelby SA, Chiang EN, Holowka DA, Baird BA. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS One 2012; 7:e31457; http://dx.doi.org/10.1371/journal.pone.0031457; PMID: 22384026
- Klein T, Löschberger A, Proppert S, Wolter S, van de Linde S, Sauer M. Live-cell dSTORM with SNAP-tag fusion proteins. Nat Methods 2011; 8:7 - 9; http://dx.doi.org/10.1038/nmeth0111-7b; PMID: 21191367
- Wombacher R, Heidbreder M, van de Linde S, Sheetz MP, Heilemann M, Cornish VW, et al. Live-cell super-resolution imaging with trimethoprim conjugates. Nat Methods 2010; 7:717 - 9; http://dx.doi.org/10.1038/nmeth.1489; PMID: 20693998
- Wolter S, Endesfelder U, van de Linde S, Heilemann M, Sauer M. Measuring localization performance of super-resolution algorithms on very active samples. Opt Express 2011; 19:7020 - 33; http://dx.doi.org/10.1364/OE.19.007020; PMID: 21503016
- Holden SJ, Uphoff S, Kapanidis AN. DAOSTORM: an algorithm for high- density super-resolution microscopy. Nat Methods 2011; 8:279 - 80; http://dx.doi.org/10.1038/nmeth0411-279; PMID: 21451515
- Huang F, Schwartz SL, Byars JM, Lidke KA. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed Opt Express 2011; 2:1377 - 93; http://dx.doi.org/10.1364/BOE.2.001377; PMID: 21559149
- Cox S, Rosten E, Monypenny J, Jovanovic-Talisman T, Burnette DT, Lippincott-Schwartz J, et al. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat Methods 2012; 9:195 - 200; http://dx.doi.org/10.1038/nmeth.1812; PMID: 22138825
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Identification of clustering artifacts in photoactivated localization microscopy. Nat Methods 2011; 8:527 - 8; http://dx.doi.org/10.1038/nmeth.1627; PMID: 21666669
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Quantitative photo activated localization microscopy: unraveling the effects of photoblinking. PLoS One 2011; 6:e22678; http://dx.doi.org/10.1371/journal.pone.0022678; PMID: 21818365
- Löschberger A, van de Linde S, Dabauvalle MC, Rieger B, Heilemann M, Krohne G, et al. Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J Cell Sci 2012; 125:570 - 5; http://dx.doi.org/10.1242/jcs.098822; PMID: 22389396
- Steinhauer C, Jungmann R, Sobey TL, Simmel FC, Tinnefeld P. DNA origami as a nanoscopic ruler for super-resolution microscopy. Angew Chem Int Ed Engl 2009; 48:8870 - 3; http://dx.doi.org/10.1002/anie.200903308; PMID: 19830751
- Williamson DJ, Owen DM, Rossy J, Magenau A, Wehrmann M, Gooding JJ, et al. Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events. Nat Immunol 2011; 12:655 - 62; http://dx.doi.org/10.1038/ni.2049; PMID: 21642986
- Owen DM, Rentero C, Rossy J, Magenau A, Williamson D, Rodriguez M, et al. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. J Biophotonics 2010; 3:446 - 54; http://dx.doi.org/10.1002/jbio.200900089; PMID: 20148419