Abstract
Described by the Belgian cytologist Christian De Duve in 1949,Citation1 lysosomes (from the Greek “digestive bodies”) are ubiquitous specialized intracellular organelles that ensure the degradation/recycling of macromolecules (proteins, lipids, membranes) through the activity of specific enzymes (i.e., acid hydrolases). They receive their substrates through different internalization pathways (i.e., endocytosis, phagocytosis and autophagy) and are involved in a wide range of physiological functions from cell death and signaling to cholesterol homeostasis and plasma membrane repair.Citation2 In Mammals, 50 soluble lysosomal hydrolases have been described, each targeting specific substrates. They are confined in the lumen of the lysosome and require an optimum pH (i.e., pH 4.5) to work. This acidic pH compared with the slightly alkaline pH of the cytosol (i.e., ~pH 7.2) is maintained by the activity of integral lysosomal membrane proteins (LMPs, that represent the second class of lysosomal proteins), including the V-type proton (H+)-ATPaseCitation3 and the chloride ion channel CLC7Citation4 that pumps protons from the cytosol across the lysosomal membrane.
To date, up to 50 lysosome-dependent diseases have been described in human (called Lysosomal Storage Diseases, LSDs)Citation5,Citation6 and are linked to mutations in the genes encoding the different lysosomal hydrolases and LMPs proteins. The Niemann-Pick (NP) diseases (first described and documented by the German physicians Albert Niemann and Ludwig Pick in 1914) are one of these rare autosomal recessive metabolic disorders that belong to the LSD family. NP diseases are classified according to their genetic origin. Hence, NP diseases type A and B are caused by mutations in the SMPD1 gene encoding the sphingomyelinase (SMase), a hydrolase involved in the catabolism of sphingomyelins (SMs) into ceramide (Cer) and phosphocholine (PC). In contrast, NP diseases type C and D are caused by mutations in the NPC1 and NPC2 genes encoding the lysosomal membrane proteins NPCs, a key element of the intracellular cholesterol/lipids trafficking. Interestingly, beside their unrelated genetic origins, NP diseases present similar cellular defects (i.e., lysosomal accumulation of free cholesterol and SMs due to insufficient SMase activity, as well as alteration of Ca2+ and Fe2+ homeostasis). Even more surprising is that another LSD, the mucolipidosis type IV (ML4), caused by mutations in the gene MCOLN1 encoding the mucolipin transient receptor potential (TRP) channel 1 (TRPML1), also display cellular defects similar to those observed in NP diseases. TRPML1 channel, which belongs to the TRP channel family,Citation7 is widely expressed and predominantly resides in the late endosomes and lysosomes,Citation8,Citation9 rendering their study difficult. It was initially proposed that TRPML1 works as a proton leak channel to prevent lysosomal over-acidification.Citation10 Further studies have then shown that TRPML1 is rather an endolysosomal Fe2+ release channel,Citation11 or even a Ca2+ and Fe2+/Mn2+ dually permeable cation channel.Citation12 Considering that lysosomal exocytosis is a Ca2+ and synaptotagmin-dependent process requiring significant cytosolic Ca2+ elevations and that the major source of Ca2+ comes from the lysosome itself (the lysosomal Ca2+ concentration has been evaluated around 0.4 to 0.6 mM),Citation13 the relative Ca2+ permeability of TRPML1 appears particularly important for lysosomal exocytosis. Although previous studies have reported TRPML1-dependent lysosomal trafficking alterations, the functional relation between TRPML1 and lysosomal accumulation of specific substrates has remained unclear. In a recent study published in Nature Communications,Citation14 the authors have examined the possible central implication of TRPML1 in the molecular physiopathology of lipid storage disorders. The results of this study indicate that SMs are potent TRPML1 inhibitors whereas SMase in contrast increases channel activity. The authors demonstrate that increasing TRPML1 activity in NP-type C cells is sufficient to restore normal lysosomal trafficking and prevent cholesterol accumulation. Therefore, primary abnormal lysosomal lipid accumulation in NP diseases (due to decrease in SMase or NPCs activity) inhibits lysosomal Ca2+ release through TRPML1 and Ca2+-dependent lysosomal trafficking that in turn causes a secondary lysosomal storage. Similarly, direct alteration of TRPML1 activity in the mucolipidosis type IV disease is sufficient to produce lysosomal storage by inhibiting lysosomal trafficking.
To carry out their studies, the authors used a combination of electrophysiological and Ca2+ fluorimetry assays to measure TRPML1 currents and cytosolic Ca2+ elevations, respectively, in a variety of NP cell lines, along with immunocytochemical and confocal imaging analysis of lysosomal trafficking and storage.
In the absence of specific available pharmacology for TRPML1 channels, the authors first performed a low-throughput screen using synthetic small molecules previously identified as TRPML3 agonists. Using whole-cell patch-clamp recording and Ca2+ fluorimetry on HEK cells overexpressing a surface-expressed TRPML1 channel (TRPML1–4A, previously identified by alanine-scanning mutagenisCitation15), they identified a compound (ML-SA1) that potently activates an inward rectifying TRPML1-dependent current similar to the one activated by phosphatidylinositol 3,5-bisphosphate (PI3,5P2), the physiological and specific TRPML1 agonist previously identified.Citation16 Although ML-SA1 is not specific for TRPML1 but also activates TRPML2 and 3 channels, it remains specific to the TRPML sub-family. Moreover, whole-endolysosome patch-clamp recordings performed on human fibroblast indicate that ML-SA1 activates a native inward rectifying current (similar to the one recorded on TRPML1–4A HEK cells) that is absent in TRPML1 deficient cells (i.e., ML4 cells derived from a ML4 patient), indicating that TRPML1 is the main endolysosomal channel at the origin of the cationic current. TRPML1 channel being an intracellular ion channel renders its study delicate in an intact cellular environment. To bypass this technical difficulty, the authors developed a TRPML1 channel fused to a Ca2+ indicator (i.e., GCaMP3). When overexpressed in HEK cells, this GCaMP3-TRPML1 channel is well targeted to the endolysosomes. Moreover, increase in fluorescence signal is mainly observed upon release of lysosomal Ca2+ (i.e., induced by osmotic lysis of the lysosomes) and not upon release of Ca2+ from the endoplasmic reticulum (i.e., induced by application of thapsigargin), demonstrating that this GCaMP3-TRPML1 Ca2+-reporter channel presents a well spatially delimited Ca2+ detection and thus appear to be an efficient molecular tool to specifically investigate TRPML1-dependent lysosomal Ca2+ release in intact cells. Furthermore, application of ML-SA1 on COS cells overexpressing GCaMP3-TRPML1 channels produce a significant Ca2+ transient that is not reproduced using a non-permeable TRPML1 channel, indicating that the cytosolic Ca2+ elevation specifically results from the release of Ca2+ from the lysosomes through TRPML1 channels. Interestingly, this Ca2+ transient is strongly reduced when GCaMP3-TRPML1 channel is expressed in NP-type C COS cells or NPC1−/− mouse macrophages (characterized by lysosomal sphingomyelins and cholesterol accumulation). Similarly, pharmacological inhibition of cholesterol transport in wild-type COS cells abolished TRPML1-dependent lysosomal Ca2+ release, indicating that lysosomal lipids accumulation inhibits TRPML1 channel activity. These results were confirmed by whole-endolysosome patch-clamp recordings performed on human NP-type C fibroblasts where the TRPML1-dependent current induced by application of ML-SA1 is strongly abolished besides a two to 3-fold increase in the TRPML1 mRNA in these cells, suggesting that lysosomal lipid accumulation tonically inhibits TRPML1 activity. Indeed, application of SMs on HEK cells expressing the surface-expressed TRPML1 channel (TRPML1–4A) strongly inhibits TRPML1-dependent current, whereas application of SMase in contrast potentiates TRPML1 activity. Similarly to what was observed in NP-type C cells, Ca2+ fluorimetry performed on NP-type A human fibroblasts expressing GCaMP3-TRPML1 channels revealed a strong decrease of the TRPML1-dependent lysosomal Ca2+ release. Finally, using confocal imaging, the authors show that abnormal lysosomal lipid accumulation observed in NPC1−/− mouse macrophages can be reversed by increasing TRPML1 activity (i.e., by application of TRPML1 agonist). Moreover, consistent with the observation that sphingomyelins constitutively inhibit TRPML1 activity, overexpression of SMase or a mutated TRPML1 channel constitutively active (TRPML1-VaCitation12,Citation17) in NP-type C CHO cells is sufficient to restore normal lysosomal cholesterol trafficking. However, the Ca2+-dependence of this process was not explored. Moreover, it is worth to mention that TRPML1 is not the unique lysosomal Ca2+ channel. Indeed, NAADP-induced lysosomal Ca2+ release relies to the activation of a two-pore channelCitation18,Citation19 and possibly TRPML1.Citation20 Future studies will undoubtedly determine the relative contribution of these two Ca2+ release pathways in lysosomal exocytosis and trafficking.
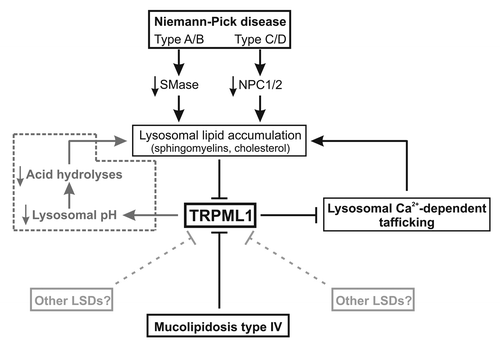
In summary, the authors have provided compelling evidences that TRPML1 channel plays a critical role in lysosomal trafficking. Abnormal lysosomal lipids accumulation due to insufficient SMase or NPCs activity in NP disease tonically inhibits TRPML1 activity that in turn inhibits lysosomal trafficking, amplifying abnormal lysosomal storage (). Interestingly, increase in TRPML1 activity is sufficient to restore lysosomal trafficking and prevent abnormal lipids accumulation. It thus appears that TRPML1 channel, possibly by controling Ca2+-dependent lysosomal trafficking, is the entry point of various lysosomal storage diseases, and therefore represents a promising pharmacological target for the treatment of lysosomal storage disorders.
Figure 1. Comprehensive signaling events mediating lysosomal storage diseases. Under normal condition, lysosomal Ca2+ release through TRPML1 channel into the cytosol supports a transient Ca2+ elevation essential for lysosomal exocytosis and lipid trafficking. Under pathological conditions, lysosomal lipid accumulation (sphingomyelins, cholesterol) caused by alteration in sphingomyelinase (SMase) and NPCs activity (i.e., Niemann-Pick disease) inhibits TRPML1 channel activity and therefore lysosomal exocytosis and trafficking that in turn cause a secondary lysosomal storage. Similarly, direct alteration of TRPML1 activity (i.e., mucolipidosis type IV) results in the same lysosomal defects. Although H+ permeability of TRPML1 channel is still controversial, alteration in TRPML1 activity could also cause lysosomal H+ accumulation and pH acidification, decreasing acid hydrolases activity and causing lysosomal storage (pathway in dotted line).
Acknowledgments
N.W is supported by a postdoctoral fellowship from Alberta Innovates Health Solutions (AIHS) and Hotchkiss Brain Institute.
References
- de Duve C. The lysosome turns fifty. Nat Cell Biol 2005; 7:847 - 9; http://dx.doi.org/10.1038/ncb0905-847; PMID: 16136179
- Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 2009; 10:623 - 35; http://dx.doi.org/10.1038/nrm2745; PMID: 19672277
- Williamson WR, Hiesinger PR. On the role of v-ATPase V0a1-dependent degradation in Alzheimer disease. Commun Integr Biol 2010; 3:604 - 7; http://dx.doi.org/10.4161/cib.3.6.13364; PMID: 21331254
- Braun AP. Identification of ClC-7 as a major pathway for Cl- movement in lysosomes. Channels (Austin) 2008; 2:309; http://dx.doi.org/10.4161/chan.2.5.7195; PMID: 18981711
- Lloyd-Evans E, Platt FM. Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011; 50:200 - 5; http://dx.doi.org/10.1016/j.ceca.2011.03.010; PMID: 21724254
- Schulze H, Sandhoff K. Lysosomal lipid storage diseases. Cold Spring Harb Perspect Biol 2011; 3:3; http://dx.doi.org/10.1101/cshperspect.a004804; PMID: 21502308
- Nilius B. TRP channels in disease. Biochim Biophys Acta 2007; 1772:805-12.
- Bach G. Mucolipin 1: endocytosis and cation channel--a review. Pflugers Arch 2005; 451:313 - 7; http://dx.doi.org/10.1007/s00424-004-1361-7; PMID: 15570434
- Colletti GA, Kiselyov K. Trpml1. Adv Exp Med Biol 2011; 704:209 - 19; http://dx.doi.org/10.1007/978-94-007-0265-3_11; PMID: 21290297
- Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, et al. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem 2006; 281:7294 - 301; http://dx.doi.org/10.1074/jbc.M508211200; PMID: 16361256
- Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008; 455:992 - 6; http://dx.doi.org/10.1038/nature07311; PMID: 18794901
- Dong XP, Wang X, Shen D, Chen S, Liu M, Wang Y, et al. Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem 2009; 284:32040 - 52; http://dx.doi.org/10.1074/jbc.M109.037184; PMID: 19638346
- Luzio JP, Bright NA, Pryor PR. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem Soc Trans 2007; 35:1088 - 91; http://dx.doi.org/10.1042/BST0351088; PMID: 17956286
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 2012; 3:731; http://dx.doi.org/10.1038/ncomms1735; PMID: 22415822
- Vergarajauregui S, Puertollano R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 2006; 7:337 - 53; http://dx.doi.org/10.1111/j.1600-0854.2006.00387.x; PMID: 16497227
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun 2010; 1:38; http://dx.doi.org/10.1038/ncomms1037; PMID: 20802798
- Grimm C, Cuajungco MP, van Aken AF, Schnee M, Jörs S, Kros CJ, et al. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc Natl Acad Sci U S A 2007; 104:19583 - 8; http://dx.doi.org/10.1073/pnas.0709846104; PMID: 18048323
- Zhu MX, Evans AM, Ma J, Parrington J, Galione A. Two-pore channels for integrative Ca signaling. Commun Integr Biol 2010; 3:12 - 7; http://dx.doi.org/10.4161/cib.3.1.9793; PMID: 20539775
- Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, et al. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J Biol Chem 2011; 286:22934 - 42; http://dx.doi.org/10.1074/jbc.M110.210930; PMID: 21540176
- Zhang F, Xu M, Han WQ, Li PL. Reconstitution of lysosomal NAADP-TRP-ML1 signaling pathway and its function in TRP-ML1(-/-) cells. Am J Physiol Cell Physiol 2011; 301:C421 - 30; http://dx.doi.org/10.1152/ajpcell.00393.2010; PMID: 21613607