Abstract
Synaptic transmission depends on several molecular and geometric components, such as the location of vesicular release, the number of released neurotransmitter molecules, the number and type of receptors, as well as the synapse organization. Our goal here is to illustrate how synaptic modeling allows extracting quantitative information in the context of neurological diseases and associated therapies. Combining electrophysiology with simulation tools, we first evaluate the reduction in synaptically released glutamate molecules induced by a ketogenic diet. In a second part, because the scaffolding molecule Shank3 is disrupted at the postsynaptic density in Autism Spectral Disorders, we present a numerical simulation of the synaptic response where this disruption leads to an alteration of the excitatory AMPA receptor trafficking. The take home message is that combining recent experimental findings with modeling approaches allows obtaining precise quantitative properties of what was still unapproachable a decade ago.
Results
Neuron communication relies on synaptic transmission, the manifestation of which is generated in the synaptic current. This current depends on several molecular and geometric components, such as the location of vesicular release, the number of released neurotransmitter molecules, the number and type of receptors, trafficking between the postsynaptic density (PSD) and extrasynaptic compartments, as well as the synapse organization. Interestingly, variations of these quantities modulate the postsynaptic current.6 The difficulty in understanding synaptic transmission relies in that it exactly stands at a scale where stochastic molecular events are integrated into a cellular function (i.e. depolarization), making a synapse an intrinsically unreliable device. This inherent difficulty is addressed by the development of mathematical models and numerical simulations based on stochastic molecular dynamics.Citation2,Citation1,Citation3,Citation4,Citation5,Citation6 Indeed, the role of modeling is precisely useful to explore any hypothesis and obtain precise quantification and predictions.
We recently presented an integrated biophysical modelCitation6 of synaptic transmission starting after neurotransmitter vesicle fusion. In the simulation of this model, each neurotransmitter molecule can be followed, as well as the number of open receptors. In addition, receptors can move freely on the postsynaptic membrane. Furthermore, glial cells unsheathing the synapse can pump neurotransmitters in and out. Using this simulation tool, we studied the statistical properties such as the mean, the variance and the coefficient of variation or any other quantities related to the postsynaptic current.Citation6 Strikingly, the most sensitive parameters in controlling the synaptic current turns out to be the co-localization of the presynaptic vesicle release sites relative to the postsynaptic receptors. The variation of this relative location leads to a continuous range of current amplitude, which may be decreased by a factor of eight during an ectopic release.Citation6 We also used this approach to interpret the larger variance exhibited by spontaneous miniature excitatory synaptic currents (mEPSCs) compared with the evoked activity (the vesicular release probability conditioned on the occurrence of an actual release event is the relevant parameter to better classify synaptic events).Citation6 Based on this, we predicted that during spontaneous activity, vesicles may be released in a larger domain at sites close and far from the postsynaptic receptors, whereas upon evoked activity, vesicle release only occurs at close sites.
To further demonstrate the advantage of our modeling approach and simulation tools, we extract here quantitative information at a molecular level. Indeed, we apply our numerical methods to two pathological examples: one concerns the ketogenic diet, well known since ancient times to offer significant anticonvulsant benefits, especially in children with severe epilepsy, and the second is Autism Spectral Disorders (ASDs), where the scaffolding molecule Shank3 is disrupted at the postsynaptic density. Our goal is to estimate: 1) the reduction in the number of released glutamate molecules induced by ketone bodies, and 2) the effect of SHANK3 mutation on glutamate receptor trafficking and on the synaptic current in ASDs.
Drop in release glutamate molecules during a ketogenic diet
Recently, a significant step in understanding the molecular basis of the ketogenic diet was achieved by observing that ketone bodies, whose amounts increase with fasting, lead to a reduction in the amplitude of spontaneous postsynaptic excitatory currents.Citation9 Indeed, vesicular glutamate transporters (VGLUTs), necessary for glutamate release by exocytosis, can be partially blocked by acetoacetate, because it competes with chloride ions at the binding sites of the transporters located on the vesicle, causing a reduction in the refilling of synaptic vesicles with glutamate molecules. Excitatory spontaneous synaptic transmission is alteredCitation9, and we show here that inducing a single stimulation in Schaffer collaterals (−C) also reduces the evoked synaptic transmission. Interestingly, the consequence of such effect is a reduction of epileptic seizures. Using our simulation tool that we recently developed,Citation6 we now estimate from the evoked and miniature electrophysiological events (ref. Citation9 and −C), the decrease in the amount of released glutamate molecules induced by the ketone bodies. For this purpose, we ran a Brownian Dynamics simulation in which a single synaptic vesicle was released at the active zone (parameters in ). Because the paired pulse ratio of synaptically-evoked currents was unchanged by ketone bodies,Citation9 the vesicle release probability is not altered, and thus it is a reduction in the number of glutamate molecules that is responsible for the decreased synaptic response. Consequently, we found that decreasing by 33% the number of glutamate molecules contained in synaptic vesicles, i.e., from 3000 to 2000 molecules (contained in a single vesicle), results in a decrease of 25% in the synaptic response, as reported in,Citation9 and in agreement with the level of depression in basal evoked synaptic transmission that we recorded in hippocampal slices (−C). Interestingly, we also found that the number of additional AMPARs that would be necessary to insert at the PSD for the synaptic current to come back to the basal response should be increased by 60% (). This result demonstrates that this would involve a mechanism as strong as long-term potentiation. In addition, this suggests that homeostatic processes resulting in a local increase in the number of AMPARs would probably not be sufficient to compensate the effect of a ketogenic diet. However, it would be interesting to investigate whether and how the decrease in synaptic transmission may reshape synaptic organization to ultimately contribute to the reduction of epileptic seizures.
Figure 1. Quantifying the effect of ketone bodies on synaptic transmission. Ketone bodies decrease basal synaptic transmission of CA1 pyramidal cells: depression of fEPSPs in (A) β-hydroxybutyrate (βHB, 11 mM) (p < 0.001, n = 8) and (B) acetoacetate (ACA, 11 mM) (p < 0.001, n = 8). Sample traces of averaged field potentials before (trace 1) and during (trace 2) ketone bodies application, as indicated by the numbers, are shown below the curves (A, B). Scale bar, 0.2 mV, 10 ms. (C) Graph summarizing the peak depressions of fEPSPs during perfusion of βHB or ACA for 30 min. (D) Schematic representation of the synapse for the simulation. (E) Brownian dynamics simulation of synaptic current response in normal (green) and after glutamate reduction (red). A small variation of the vesicular release in the active zone has little effect (dotted red). (F) Effect of increasing the number of AMPARs, necessary to compensate for the reduction in the number of released glutamate molecules.
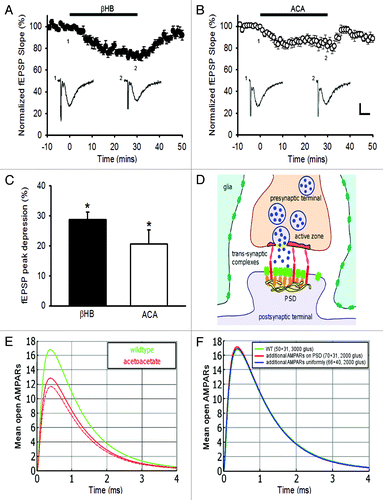
Table 1. Simulation parameters for the synaptic current
Thus we conclude that under ketone bodies, a reduction of 33% in the number of glutamate molecules released from synaptic vesicles leads to a decrease in the synaptic response of about 25%. In addition, it is very unlikely that this decrease may be compensated by insertion of AMPARs at the PSD, because this would require adding a high amount of receptors.
Autism spectrum disorders: Simulation of the synaptic response in a numerical setting mimicking SHANK3 mutation
In the second example, we propose to explore the alteration of synaptic transmission in a model scenario for Autism Spectrum Disorders, which was recently shown to be associated to a mutation in the Shank3 scaffolding protein,Citation8 a fundamental molecule of the PSD interacting with PSD 95. Shank3 is located at the tip of actin filaments and enhances its polymerization. A SHANK3 mutation affects the morphology of dendritic spines, and was shown to reduce synaptic transmission in mature neurons.Citation10,Citation11
Since Shank3 proteins form a key postsynaptic scaffold at glutamatergic synapses and interacts with many synaptic proteins, including the neuroligin-neurexin complex, we hypothesize that the mutation disturbs receptor trafficking and stabilization at the PSD.Citation11 We thus applied our simulation toolCitation6 to a synapse in which vesicle release sites were not co-localized with AMPARs. To account for the alteration, we distributed the AMPARs uniformly over the postsynaptic terminal, rather than concentrating them at the PSD. Based on the disruption of the trans-synaptic neuroligin-neurexin complexes, we further distributed the vesicular release sites uniformly over the active zone (blue, ), and then uniformly over the presynaptic terminal (dashed blue). We found that under these modifications, the amplitude of the synaptic current is reduced by 40% ().
Figure 2. Quantifying the effect of Shank3-like mutation on synaptic transmission. (A) Schematic representation of the synapse with neuroligin and Shank3 disruption. (B) Synaptic response in normal (green) and Shank3-like mutation (blue) upon spreading the vesicle release sites, distributed as in a wild type synapse (solid blue) and over the pre-synaptic terminal (dotted). AMPARs are uniformly distributed over the postsynaptic terminal (blue) and vesicles are distributed over the presynaptic terminal (dashed blue).
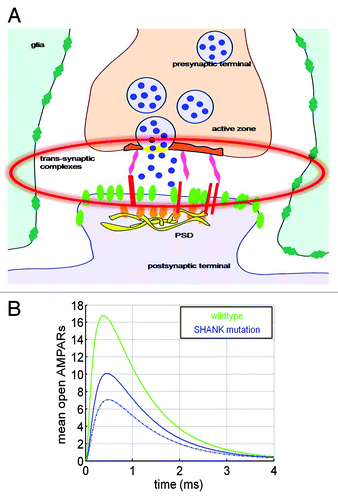
This scenario suggests that indeed decrease in the synaptic current can be due to a misalignment of the vesicular release with the PSD. In addition, the current is further reduced, since AMPARs are not concentrated at the PSD, leading to a decrease in the probability of opening a large number of them.
At this stage, we have shown that spreading vesicle release sites and postsynaptic receptors, effectively removing their co-localization, leads to a decrease in the synaptic current. Other consequences should be further explored, for example on the trafficking of AMPARs at the PSD and their associated residence time.
Discussion and Conclusions
We have first quantified here the reduction in the number of released glutamate molecules during synaptic events in the presence of ketone bodies. We report here that a 33% reduction leads to a decrease in the synaptic response of about 25% in the current amplitude. We have further shown that this reduction cannot be compensated by increasing AMPARs at the PSD, which would require a large number of additional receptors, that are usually produced and trafficked during synaptic plasticity stimulations.Citation15 This reduction in the number of released glutamate molecules can ultimately change the steady-state of the neural network, leading to a decrease in field excitatory postsynaptic potentials, noise amplitude and frequency of the spontaneous burst discharges. Unfortunately, breaking the ketogenic diet with even the smallest ingestion of sugar leads rapidly to the re-appearance of epileptic seizures, suggesting that no permanent changes underlying the decreased strength of synaptic transmission, such as a reorganization of neuronal connectivity, occurs. Surprisingly homeostatic mechanisms, consisting for instance in increasing the number of postsynaptic AMPA receptors, do not operate to counteract the decreased number of released glutamate molecules. This may be due to the limiting energy substrates available during the ketogenic diet, which are incompatible with the costly insertion of AMPA receptors at the postsynaptic density. This situation is in contrast with the permanent and stable reorganization of synapses, associated with a change from epileptic to a normal regime in certain brain regions, such as the auditory cortex.Citation16 Indeed combinations of experimental and modeling approaches have shown that decreasing synaptic efficacy during specific learning tasks leads to a permanent change in cortical maps, neural network connectivity and activity.Citation16 To conclude, although the ketogenic diet leads to a decrease in the synaptic current, it does not lead to any significant permanent changes in the synaptic strength which could remodel the network.
In the second study, we used a computational model to disrupt the interaction between scaffolding proteins and AMPARs. As a result we obtained a 40% reduction in the amplitude of the synaptic current, consistent with a decrease in basal excitatory synaptic strength reported in Neurexin-1-KO mice.Citation13 Although we hypothesized here that the reduction in synaptic transmission is due to the misalignment of vesicular release and the absence of cluster of receptors, we cannot rule out an additional effect associated with a geometric reorganization in neuroglial cell interactions,Citation8 which would directly result in a fast and massive glutamate uptake.Citation8
As illustrated here by two different examples, biophysical modeling and numerical simulation studies can now be used to test specific assumptions and obtain precise quantification of synaptic transmission in diseases that involve alterations in synaptic functions.
Materials and Methods
Electrophysiology
Acute transverse hippocampal slices (400 μm) were prepared as previously describedCitation17 from 16 to 25 d-old wild-type C57BL6 mice. For all analyses, mice of both genders and littermates were used. Slices were maintained at room temperature in a storage chamber containing an artificial cerebrospinal fluid (ACSF) (in mM: 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, saturated with 95% O2 and 5% CO2) for at least 1 h before recording. Slices were transferred to a submerged recording chamber mounted on an Olympus BX51WI microscope and were perfused with ACSF at a rate of 1.5 ml/min at room temperature. Extracellular field recordings were performed. Evoked postsynaptic responses were induced by stimulating Schaffer collaterals (0.1 Hz) in CA1 stratum radiatum with ACSF filled glass pipettes. Field excitatory postsynaptic potentials (fEPSPs) were recorded with glass pipettes (2–5 MΩ) filled with ACSF and placed in stratum radiatum. Recordings were acquired with Multiclamp-700B amplifier (Molecular Devices), digitized at 10 kHz, filtered at 2 kHz, stored and analyzed on computer using pClamp 10 and Clampfit 10 softwares (Molecular Devices). All data are expressed as mean ± SEM. Statistical significance for comparisons was determined by Student’s paired t-test. β-hydroxybutyrate (βHB) and Li-acetoacetate (ACA) were obtained from Sigma.
Modeling and simulations
Simulations were conducted as described in reference Citation6. For details see also Supplemental Information of reference Citation6. The default values for all parameters are listed in . For our simulations, a wildtype synapse is modeled with presynaptic and postsynaptic terminals which are two coaxial cylinders of 800 nm diameter. These cylinders were placed at a distance of 20 nm to represent the synaptic cleft. The glial sheet was modeled by a coaxial cylindrical surface laterally surrounding the cylinders at a distance of 40 nm. The PSD was defined as a disk of 300 nm diameter centered on the surface of the postsynaptic cylinder (see ). Vesicle release sites were located in the center of the intra-cleft surface of the presynaptic cylinder. A single vesicle contained 3000 glutamate molecules. Upon vesicle fusion, they were all released at a single point and in a single time step.
Glutamate molecules could diffuse freely with a diffusion constant of 0.2 µm2/sCitation1 and their motion was simulated by Brownian dynamics. Upon hitting a membrane surface, trajectories were reflected (or bound on transporters). Upon reaching a distance of 0.5 µm away from the cleft center, trajectories were terminated. The PSD contained 50 AMPARs and the remaining intra-cleft surface contained 31 AMPARs corresponding to 10% of the receptor density on the PSD. The receptor kinetics was modeled by a Markov scheme.Citation6,Citation14 The glial sheath was uniformly covered with glutamate transporters, which were located on an equally-spaced lattice at a density of 5 000 per µm2. The kinetics of the transporters involved binding, internalizing, and unbinding glutamate molecules and was modeled by a Markov scheme.Citation1
In the model for the ketogenic diet, the vesicle load was reduced to 2 000 molecules. The SHANK-KO synapse model deviated from the wildtype synapse by the following: Vesicle release sites were uniformly distributed on the intra-cleft surface of the presynaptic cylinder and AMPAR receptors were placed inside and outside the PSD at equal densities.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Franks KM, Stevens CF, Sejnowski TJ. Independent sources of quantal variability at single glutamatergic synapses. J Neurosci 2003; 23:3186 - 95; PMID: 12716926
- Holmes WR. Modeling the effect of glutamate diffusion and uptake on NMDA and non-NMDA receptor saturation. Biophys J 1995; 69:1734 - 47; http://dx.doi.org/10.1016/S0006-3495(95)80043-3; PMID: 8580317
- Barbour B. An evaluation of synapse independence. J Neurosci 2001; 21:7969 - 84; PMID: 11588170
- Holcman D. Computational challenges in synaptic transmission, AMS Contemporary Mathematics (CONM) book series. Mathematical and Computational Challenges 2009; 494:1 - 26
- Taflia A, Holcman D. Estimating the synaptic current in a multi-conductance AMPA receptor model. Biophysical Journal. Biophys J. 2011; 101:781 - 92
- Freche D, Pannasch U, Rouach N, Holcman D. Synapse geometry and receptor dynamics modulate synaptic strength. PLoS One 2011; 6:e25122; http://dx.doi.org/10.1371/journal.pone.0025122; PMID: 21984900
- Blanpied TA, Kerr JM, Ehlers MD. Structural plasticity with preserved topology in the postsynaptic protein network. Proc Natl Acad Sci U S A 2008; 105:12587 - 92; http://dx.doi.org/10.1073/pnas.0711669105; PMID: 18723686
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008; 455:903 - 11; http://dx.doi.org/10.1038/nature07456; PMID: 18923512
- Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, et al. Metabolic control of vesicular glutamate transport and release. Neuron 2010; 68:99 - 112; http://dx.doi.org/10.1016/j.neuron.2010.09.002; PMID: 20920794
- Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 2012; 17:71 - 84; http://dx.doi.org/10.1038/mp.2011.57; PMID: 21606927
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011; 472:437 - 42; http://dx.doi.org/10.1038/nature09965; PMID: 21423165
- Etherton MR, Tabuchi K, Sharma M, Ko J, Südhof TC. An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J 2011; 30:2908 - 19; http://dx.doi.org/10.1038/emboj.2011.182; PMID: 21642956
- Etherton MR, Blaiss CA, Powell CM, Südhof TC. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A 2009; 106:17998 - 8003; http://dx.doi.org/10.1073/pnas.0910297106; PMID: 19822762
- Milstein AD, Zhou W, Karimzadegan S, Bredt DS, Nicoll RA. TARP subtypes differentially and dose-dependently control synaptic AMPA receptor gating. Neuron 2007; 55:905 - 18; http://dx.doi.org/10.1016/j.neuron.2007.08.022; PMID: 17880894
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron 2003; 40:361 - 79; http://dx.doi.org/10.1016/S0896-6273(03)00640-8; PMID: 14556714
- Bart E, Bao S, Holcman D. Modeling the spontaneous activity of the auditory cortex. J Comput Neurosci 2005; 19:357 - 78; http://dx.doi.org/10.1007/s10827-005-3099-4; PMID: 16502241
- Pannasch U, Vargová L, Reingruber J, Ezan P, Holcman D, Giaume C, et al. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A 2011; 108:8467 - 72; http://dx.doi.org/10.1073/pnas.1016650108; PMID: 21536893