Abstract
Neurons have unique challenges relative to other cell types. Unlike most other cells, neurons must remain healthy and functional throughout the lifespan of an animal. Premature neuronal loss underlies many age-related neurodegenerative diseases, including Alzheimer and Parkinson Diseases. Despite previous research aimed at understanding the mechanisms of age-related neurodegenerative diseases, little is known about the mechanisms that allow neurons to remain functional for the lifetime of a healthy animal. Understanding these cellular and biochemical processes is essential to promote healthful aging and reduce the severity of neurodegenerative disease. Here we discuss our recent identification of neuron-specific proteins that regulate endosome fusion events and the role of endosomes in maintaining healthy neurons.
Unlike most cells of multicellular organisms, neurons cannot be replenished. A particular neuron’s network of multicellular connections cannot be easily replicated by a new cell. Since neuronal death represents a permanent loss of function for an animal, neurons have been specialized to promote their health and functionality for as long as the organism survives. We call this characteristic neuronal longevity. Neurons actively resist and repair cellular damage, and dysfunction of these processes leads to neuronal degeneration.Citation1
One process which promotes neuronal longevity is macroautophagy, herein referred to as autophagy.Citation2 Autophagy is the process by which cells degrade their own cytoplasm and organelles, either in response to starvation or to remove defective and long-lived components.Citation3,Citation4 Autophagy begins with the formation of an phagophore, which encircles the material to be degraded and then fuses with itself to form a double-membraned autophagosome. Autophagosomes fuse with endosomes and lysosomes to acquire the proteases and acidification machinery required for degradation. As the autophagosome becomes acidified, it matures into an autolysosome and completes the degradation of its contents.
Multiple lines of evidence have shown that autophagy promotes neuronal longevity. Inhibition of autophagy causes neurons to degenerate in both flies and mice.Citation5-Citation7 Interestingly, emerging evidence has demonstrated accumulation of defective autophagosomes and undegraded protein aggregates in many neurodegenerative diseases, including Alzheimer disease (AD), Huntington disease (HD), and Parkinson disease (PD. Moreover, treatment of mouse models of AD, HD, and PD with rapamycin, which induces high levels of autophagy, results in the clearance of aggregates and the reduction of disease symptoms.Citation8 Rapamycin treatment also rescues neurodegeneration in in vivo mouse models of progeria and ischemia, and in vitro models of oxygen or glucose deprivation.Citation9,Citation10 Thus, autophagy keeps healthy neurons from degenerating and protects neurons from damage and disease.
Autophagy occurs in all cells, but is enhanced in neurons to promote neuronal longevity. Neurons have higher autophagic flux than other cells, meaning that autophagosomes mature and degrade their contents faster in neurons than in other cells.Citation11 Two recent reports have found constitutive formation and retrograde transport of autophagic structures down the axons of live neurons.Citation12,Citation13 Photobleaching experiments demonstrated that autophagosomes are formed in terminals but not axons.Citation13 However, these autophagosomes are not acidified in terminals, but become acidified during retrograde transport.Citation13 These data suggest that autophagosomes fuse with endosomes or lysosomes before entering the axon and mature as they move toward the soma.
Such neuron-specific regulation of autophagy requires the presence of neuron-specific regulatory factors. We recently identified neuronal synaptobrevin (n-syb) as such a factor in Drosophila melanogaster.Citation14 N-syb, known as Synaptobrevin or VAMP2 in mice and humans, is a SNARE protein that has long been known to mediate the fusion of synaptic vesicles with the presynaptic membrane to allow neurotransmitter release.Citation15 N-syb binds the SNAREs syntaxin1 (Syx1) and SNAP-25 on the presynaptic membrane to cause synaptic vesicle fusion. Loss of n-syb function through mutation or cleavage by tetanus toxin blocks synaptic vesicle fusion and neurotransmission. We found that n-syb mutant photoreceptor neurons degenerate over time.Citation14 Over five weeks, more than half of the photoreceptors in n-syb eyes degenerated. This degeneration was due to loss of n-syb function, as photoreceptor loss was rescued by the expression of an n-syb cDNA. Other mutations that block synaptic vesicle fusion or neurotransmission did not cause degeneration, indicating that degeneration is due to an unappreciated function of n-syb.
To identify this novel function, we first characterized the subcellular localization of n-syb protein. In the nerve terminal, n-Syb not only colocalized with synaptic vesicle markers but also with early endosome markers Rab5 and Syx7. In n-syb mutant neurons, endosome markers accumulated up to four times wild-type levels. N-syb nerve terminals had high levels of proteins endocytosed from the plasma membrane. In fact, n-syb terminals swelled due to the amount of material they accumulated. Ultrastructural analysis showed that n-syb terminals were packed with small vesicles and autophagosomes. Importantly, autophagosomes were present at much higher levels than typically seen in neurons, and many already contained electron-dense material, indicating premature acidification and initiation of protein degradation. We concluded that n-Syb has a previously unappreciated role regulating endosomes and autophagosomes.
Since n-syb did not colocalize with autophagosomes, the autophagosome defects in n-syb neurons were likely a secondary effect of n-syb’s endosomal function. Autophagosomes fuse with endosomes or lysosomes to mature, so a defect in endosomal function can lead to non-maturing or slowly maturing autophagosomes. To assay endosomal function, we measured the maturation of the protease Cathepsin L. Cathepsins are synthesized as pro-proteins with an inhibitory domain blocking protease function. Pro-Cathepsins are transported from the Golgi apparatus to early endosomes, which then mature into late endosomes and lysosomes. As endosomes mature and become acidic, the Cathepsin pro-domains are cleaved off to activate protease function.Citation16 N-syb neurons accumulate high levels of pro- and mature-Cathepsin L. This data suggest that n-syb neurons accumulate early, non-acidified, endosomal compartments as well as maturing, acidified autophagosomes. We also found that pro-Cathepsin L accumulated before mature Cathepsin L during pupal development, indicating that endosomal dysfunction preceded autophagosome accumulation.
N-syb is not the only neuron-specific gene that regulates the endolysosomal system. Our previous work showed that v100, a neuron-specific subunit of the vacuolar ATP-ase, is required for the proper maturation of endosomes and autophagosomes in neurons.Citation17 The vacuolar ATP-ase acidifies endocytic organelles, but v100 has an acidification-independent function regulating SNARE-mediated membrane fusion. V100 localizes to synaptic vesicles and endosomes, regulating membrane fusion through direct interaction with Syx1 on synaptic vesicles and Syx7 on early endosomes.Citation17,Citation18 Neurons mutant for v100 accumulate synaptic vesicles, endosomes, and autophagosomes in a manner similar to n-syb mutants and they also degenerate over time.
Further analysis strengthened the connection between n-syb and v100. N-syb photoreceptors failed to localize v100 to synapses. Moreover, overexpression of a v100 cDNA in n-syb photoreceptors significantly rescued photoreceptor degeneration. Notably, a v100 cDNA carrying a mutation that blocks its acidification function in the V-ATPase still rescued degeneration as well as the wild-type v100 cDNA. Therefore, our work implicates both synaptobrevins and syntaxins in regulating endosome fusion to promote neuronal longevity.
SNAP-25, the third SNARE required for membrane fusion, may also protect against neurodegeneration. CSPα is a neuron-specific molecular chaperone whose loss causes neurodegeneration in flies and mice.Citation19 A recent study found that degeneration in murine CSPα neurons is primarily caused by loss of functional SNAP-25.Citation20 Thus, three neuron-specific genes regulate membrane fusion: n-syb, v100, and CSPα (). These genes are required for fusion of synaptic vesicles with the presynaptic membrane, and their loss causes degeneration with an accumulation of autophagosomes. We have now shown that n-syb and v100 both have important roles in endosome function. An endosomal role for CSPα has not been investigated to our knowledge.
Figure 1. A model for endosomal regulation of neuronal longevity. (A) Neuron-specific factors regulate endosome-vesicle fusion. The SNARE proteins n-Syb, SNAP-25, and Syx7 directly control membrane fusion between neuronal endosomes and vesicles. CSPα is a chaperone that maintains the stability of SNAP-25. V100 binds to Syx7 and is required for fusion through a currently unclear mechanism. Neuron-specific proteins are gray. (B) Endosomes are required for the maturation of neuronal autophagosomes. Phagophores form and engulf cytoplasm, becoming autophagosomes, in nerve terminals. In order to mature, autophagosomes first fuse with endosomes, acquiring proteases and acidification machinery. Then, autophagosomes enter the axon and begin retrograde transport to the soma. As they move, autophagosomes begin to degrade their contents (degraded material is shown black material inside the autophagosome).
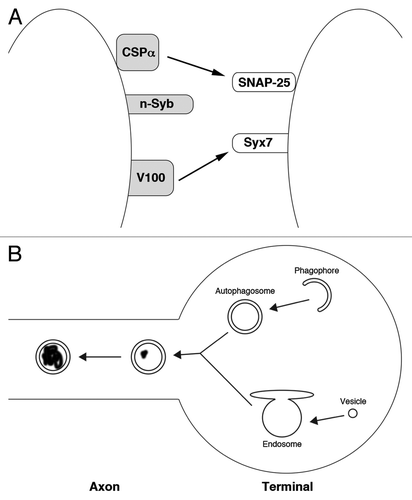
We propose that the following mechanism promotes neuronal longevity (): autophagosomes constitutively form at nerve terminal to degrade old and damaged proteins. These autophagosomes then fuse with endosomes before entering the axon. Endosomal V-ATPases and proteases cause the maturation of these autophagosomes into autolysosomes as they move down the axon. Therefore, any mutation or treatment that interferes with endosome protease function blocks autophagosome transport and maturation. Autophagosome accumulation would induce neuronal stress, leading to cell death. Thus, regulation of endosomes by neuron-specific factors is essential for proper autophagy and neuronal longevity.
| Abbreviations: | ||
| AD | = | Alzheimer Disease |
| HD | = | Huntington Disease |
| PD | = | Parkinson Disease |
| n-syb | = | neuronal synaptobrevin |
| syx1 | = | syntaxin1 |
Acknowledgments
The authors thanks Tracie Paine, Gunnar Kwakye and Robin Hiesinger for comments on this manuscript.
References
- Heiman MG, Pallanck L. Neurons at the extremes of cell biology. Mol Biol Cell 2011; 22:721; http://dx.doi.org/10.1091/mbc.E10-12-0965; PMID: 21406584
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Meléndez A, Neufeld TP. The cell biology of autophagy in metazoans: a developing story. Development 2008; 135:2347 - 60; http://dx.doi.org/10.1242/dev.016105; PMID: 18567846
- Wang CW, Klionsky DJ, Klionsky DJ. The molecular mechanism of autophagy. Mol Med 2003; 9:65 - 76; PMID: 12865942
- Wang T, Lao U, Edgar BA. TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol 2009; 186:703 - 11; http://dx.doi.org/10.1083/jcb.200904090; PMID: 19720874
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Lee, J.-A. Autophagy in neurodegeneration: two sides of the same coin. BMB reports 42, 324-30 (2009).
- Mariño G, Fernández AF, López-Otín C. Autophagy and aging: lessons from progeria models. Adv Exp Med Biol 2010; 694:61 - 8; http://dx.doi.org/10.1007/978-1-4419-7002-2_6; PMID: 20886757
- Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 2012; 8:77 - 87; http://dx.doi.org/10.4161/auto.8.1.18274; PMID: 22113203
- Boland, B. et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 6926-37 (2008).
- Lee, S., Sato, Y. & Nixon, R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 7817-30 (2011).
- Maday S, Wallace KE, Holzbaur ELF. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 2012; 196:407 - 17; http://dx.doi.org/10.1083/jcb.201106120; PMID: 22331844
- Haberman A, Williamson WR, Epstein D, Wang D, Rina S, Meinertzhagen IA, et al. The synaptic vesicle SNARE neuronal Synaptobrevin promotes endolysosomal degradation and prevents neurodegeneration. J Cell Biol 2012; 196:261 - 76; http://dx.doi.org/10.1083/jcb.201108088; PMID: 22270918
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science 2009; 323:474 - 7; http://dx.doi.org/10.1126/science.1161748; PMID: 19164740
- Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresić K, et al. Apoptotic pathways: involvement of lysosomal proteases. Biol Chem 2002; 383:1035 - 44; http://dx.doi.org/10.1515/BC.2002.112; PMID: 12437086
- Williamson WR, Wang D, Haberman AS, Hiesinger PR. A dual function of V0-ATPase a1 provides an endolysosomal degradation mechanism in Drosophila melanogaster photoreceptors. J Cell Biol 2010; 189:885 - 99; http://dx.doi.org/10.1083/jcb.201003062; PMID: 20513768
- Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 2005; 121:607 - 20; http://dx.doi.org/10.1016/j.cell.2005.03.012; PMID: 15907473
- Chamberlain LH, Burgoyne RD. Cysteine-string protein: the chaperone at the synapse. J Neurochem 2000; 74:1781 - 9; http://dx.doi.org/10.1046/j.1471-4159.2000.0741781.x; PMID: 10800920
- Sharma M, et al. CSPα knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J 2012; 31:829 - 41; http://dx.doi.org/10.1038/emboj.2011.467