Abstract
Our recent identification of an exosomal route for tau protein secretion1 marks a key similarity between tau and other aggregation-prone proteins implicated in neurodegenerative disease pathogenesis and is to some extent congruent with the popular idea that tau pathology spreads between neurons via a “prionlike” template-mediated protein misfolding mechanism in AD and other tauopathies. However, the observation that much of the phosphotau in CSF samples from early AD patients is exosomal (and thus likely to have been secreted) calls into question a very widely held and plausible assumption - the idea that the elevated CSF-tau in AD is due to the passive release and accumulation of tau in the CSF as a consequence of widespread neuronal death. Here we examine this issue directly and explore some of the broader implications of this study for our understanding of AD pathogenesis and the prospects for improving its diagnosis and treatment.
While significant progress has been made toward understanding the role played by tau misprocessing in AD pathogenesis, much remains unclear. At the cellular level of analysis, misprocessing events such as oligomerization, hyperphosphorylation and cleavage that lead to the tau aggregation via its microtubule binding repeat (MTBR) region have been linked to many aspects of tau mediated toxicity, especially in “tau-only” tauopathies associated with exonic point mutations in tau.Citation2-Citation4 However, tau is now known to have many cellular functions beyond its classic role in stabilizing axonal microtubules via the MTBR,Citation5,Citation6 including interactions with signal transduction and (now) unconventional secretory pathways, some of which are implicated in AD and non-AD tauopathy pathogenesis.Citation7 The classic view of a single MTBR-mediated tau toxicity mechanism in AD has been complicated over the past decade by numerous reports of an alternative toxicity mechanism that does not require the tau MTBR Citation8-Citation11 and which mediates much of the neurotoxicity due to Abeta in AD.Citation12,Citation13 The existence of separate MTBR+ and MTBR- tau toxicity mechanisms is particularly relevant to our emerging understanding of interneuronal aspects of tauopathy pathogenesis, which until very recently was based solely on neuropathology patterns suggestive of paracellular and trans-synaptic lesion propagation in AD.Citation14-Citation16 More recently, we and others have characterized tau secretion, uptake and extracellular toxicity in various cell culture modelsCitation1,Citation17-Citation20 and have shown that tau can be secreted from and taken up by non-moribund neurons in situ in a cell-autonomous non-transgenic model,Citation18,Citation21,Citation22 and that tau misprocessing and secretion resembles that seen in other aggregate-associated diseases, including prion diseases.Citation23-Citation25 These studies emphasize both the likely relevance of tau secretion to tauopathy pathogenesis and the multiplicity of possible tau transfer and toxicity pathways. While recent studies in murine transgenic models suggest the operation of a “prionlike” mechanism of lesion spread via templated protein misfolding,Citation26-Citation28 the possible involvement of MTBR- and/or receptor-mediated interneuronal toxicity mechanisms indicate that the identification of any one mechanism as responsible for lesion propagation in tauopathies is premature, particularly AD.Citation9,Citation29-Citation31 Thus, while we now have much more evidence that tau protein transfer between neurons is important to tau lesion propagation in tauopathies, the central mechanistic features of such transfer remain to be worked out. This is also true for the mechanisms by which both N-terminal (MTBR-) and near full length (MTBR+) tau species are generated in both the extracellular space and CSF.Citation32-Citation37
Is neuron death a necessary preliminary to the generation of CSF tau in AD?
One of the more important effects of our recent study may be to call in to question a widely held assumption about the significance of CSF-tau in AD pathogenesis –that elevated CSF-tau levels typically seen in AD are caused by passive release of tau from dead neurons.Citation1 Even in the earliest accounts of elevated CSF-tau in AD in the mid 1990s, this assumption was made without supporting citations or discussion of alternative mechanisms.Citation38,Citation39 The death-based origin for CSF-tau was generally accepted at that time because it was plausible, consistent with the known progression of neurofibrillary degeneration to extracellular “ghost tangles” and was unopposed by other explanations.Citation40,Citation41 Also, the lack of tauopathy models and of accurate data on the timecourse of neuron death in AD made acquiring direct evidence for alternative CSF-tau biogenesis mechanisms impractical.Citation6,Citation42 Since then, this assumption has been repeatedly (if casually) asserted in the medical literature despite the lack of evidence for a causal link between antecedent neuronal cell death and elevated CSF-tau levels in AD.
The continued plausibility of the death-induced hypothesis of CSF-tau in AD appears to be based on analogy with episodic conditions (head trauma, stroke, severe seizures) in which the time course of CSF or blood tau levels can be measured relative to a single generative event. In each of these conditions, CSF and/or serum tau levels undergo a large transient rise that is directly correlated with both event severity and with direct measures of neuronal loss.Citation43-Citation45 The analogy with AD-induced neuron loss is strengthened by the presence of excitotoxic features in the neuron loss in all of these conditions, although this does not rule out a role for active secretion, since Ca2+ fluxes also play a critical role in most unconventional secretion mechanismsCitation46. Another situation where neuron death remains a highly plausible (if still unproven) source for elevated CSF-tau may be prion diseases such as Creutzfeldt Jacob Disease (CJD), which typically features much higher CSF-tau levels than does AD together with massive neocortical neuron loss over a shorter timecourse after diagnosis.Citation47
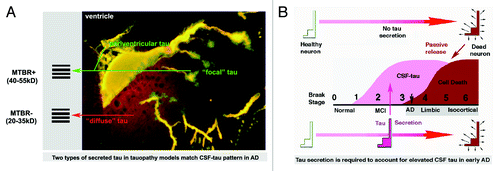
In the case of AD, however, indications from studies performed in the past decade have generally been either ambiguous or inconsistent with the “death tau” hypothesis.Citation48,Citation49 The advent of quantitative ELISA-based studies comparing CSF-tau levels in early and late AD cases (including ours) show that the well-established neuropathological “Braak” sequence of AD development does not anticipate the elevation of CSF-tau levels and thus conflicts with the death origin hypothesis (see ).Citation1,Citation14,Citation32 This is especially notable in early “limbic” stage AD (Braak Stages 3–4), when neurofibrillary pathology is confined to limbic regions of the temporal lobe that represent less than 10% of brain volume, while CSF-tau levels are rising sharply to their maximal levels.Citation1 Moreover, CSF-tau levels remain largely stable or may even fall in late stage AD, even as neurofibrillary pathology and neuron loss become widespread in the brain.Citation1,Citation14,Citation32 In this context, our demonstration that tau, particularly in its phosphorylated form, is associated with a secretion marker in CSF from early AD patients provides an alternative mechanism to neuron death, and thus may prompt a broad reassessment of the origin of CSF tau in AD.
Figure 1. Elevated CSF levels of total tau and phosphotau in early AD are better explained by secretion of misprocessed tau from neurons and glia rather than a consequence of massive neuronal death. (A) CSF-tau in AD consists largely of 1) N-terminal fragments between 20–35kD apparent molecular weight with a variable admixture of higher MW species that appear to represent near full length tau. These match the secreted tau species seen in both in situ and cell culture models of tau secretion. The image at right shows an identified neuron (ABC) in the lamprey brain expressing 4R0N human tau with the P301L tauopathy mutation after 20 d of expression immunolabeled with tau12 (N-terminal mAb – red channel) and the GFP tag (green channel). This image illustrates the multiple possible secretion routes for tau that ultimately accumulates in the CSF. The “diffuse” tau described in the lamprey model consists largely of N-terminal fragments that lack the MTBR, whereas the “focal” route requires the presence of the MTBR. Both secretion routes in the lamprey model either introduce tau to the interior surfaces of the IVth ventricle (periventricular tau) or cross it entirely. (B) The respective time courses of neuronal death and CSF-tau elevation in AD are inconsistent with postmortem passive leakage of tau into the CSF, with the highest levels of CSF-tau occurring well before the onset of widespread cerebral occurrence of neurofibrillary degeneration in the so-called isocortical stages (Braak 5–6) of AD, and failing to increase with disease severity.
The likelihood that tau secretion is involved in the genesis of CSF tau is fundamentally important to critical questions relating to AD diagnosis and treatment, since it raises broad issues of the timing and distribution of degenerative changes in the brain. For instance, a “death” mechanism of CSF-tau pathogenesis implies that tau misprocessing is fatal to affected neurons earlier in the disease than is suggested by a “secretion” based mechanism. It therefore suggests a more pessimistic outlook than the latter for the development of prospective diagnostics and effective therapeutics for AD. It is a truism that the staying power of powerful and compelling ideas is better linked to their plausibility rather than to the actual evidence supporting them – a well known example being the miasmatic theory of infectious disease.Citation50 By presenting the first direct evidence for an alternative mechanism, this study may crystallize the existing evidence that antecedent neuron death is not currently a plausible mechanism for CSF-tau biogenesis in AD. This provides a new perspective on AD pathogenesis that opens unexplored avenues to improving both the diagnosis and treatment of this devastating and widespread condition.
| Abbreviations: | ||
| AD | = | Alzheimer’s Disease |
| Abeta | = | beta amyloid peptide |
| CNS | = | central nervous system |
| CSF | = | cerebrospinal fluid |
| 4R0N | = | tau isoform with 4 microtubule binding repeats and no N terminal inserts |
| Exon 2,3 | = | tau lacking N terminal inserts |
| MTBR | = | microtubule binding repeat |
| CJD | = | Creutzfeldt Jacob Disease |
References
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 2012; 287:3842 - 9; http://dx.doi.org/10.1074/jbc.M111.277061; PMID: 22057275
- Delacourte A, David JP, Sergeant N, Buée L, Wattez A, Vermersch P, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999; 52:1158 - 65; http://dx.doi.org/10.1212/WNL.52.6.1158; PMID: 10214737
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 2001; 24:1121 - 59; http://dx.doi.org/10.1146/annurev.neuro.24.1.1121; PMID: 11520930
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta 2005; 1739:240 - 50; http://dx.doi.org/10.1016/j.bbadis.2004.08.007; PMID: 15615642
- Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990; 343:461 - 3; http://dx.doi.org/10.1038/343461a0; PMID: 2105469
- Hall GF. The biology and pathobiology of tau protein. In: The Cytoskeleton and Human Disease. Kavallaris M (ed), Springer Verlag, 2012; In press.
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, et al. Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci 2004; 24:2304 - 12; http://dx.doi.org/10.1523/JNEUROSCI.4162-03.2004; PMID: 14999081
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 2005; 25:5365 - 75; http://dx.doi.org/10.1523/JNEUROSCI.1125-05.2005; PMID: 15930385
- Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A 2006; 103:2892 - 7; http://dx.doi.org/10.1073/pnas.0511065103; PMID: 16477009
- Calissano P, Matrone C, Amadoro G. Apoptosis and in vitro Alzheimer disease neuronal models. Commun Integr Biol 2009; 2:163 - 9; PMID: 19513272
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010; 142:387 - 97; http://dx.doi.org/10.1016/j.cell.2010.06.036; PMID: 20655099
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol 2006; 175:541 - 6; http://dx.doi.org/10.1083/jcb.200605187; PMID: 17101697
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 2002; 99:6364 - 9; http://dx.doi.org/10.1073/pnas.092136199; PMID: 11959919
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82:239 - 59; http://dx.doi.org/10.1007/BF00308809; PMID: 1759558
- Su JH, Deng G, Cotman CW. Transneuronal degeneration in the spread of Alzheimer’s disease pathology: immunohistochemical evidence for the transmission of tau hyperphosphorylation. Neurobiol Dis 1997; 4:365 - 75; http://dx.doi.org/10.1006/nbdi.1997.0164; PMID: 9440125
- Armstrong RA, Cairns NJ, Lantos PL. What does the study of the spatial patterns of pathological lesions tell us about the pathogenesis of neurodegenerative disorders?. Neuropathology 2001; 21:1 - 12; http://dx.doi.org/10.1046/j.1440-1789.2001.00373.x; PMID: 11304036
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009; 284:12845 - 52; http://dx.doi.org/10.1074/jbc.M808759200; PMID: 19282288
- Kim W, Lee S, Hall GF. Secretion of human tau fragments resembling CSF-tau in Alzheimer’s disease is modulated by the presence of the exon 2 insert. FEBS Lett 2010; 584:3085 - 8; http://dx.doi.org/10.1016/j.febslet.2010.05.042; PMID: 20553717
- Kim W, Lee S, Jung C, Ahmed A, Lee G, Hall GF. Interneuronal transfer of human tau between Lamprey central neurons in situ. J Alzheimers Dis 2010; 19:647 - 64; PMID: 20110609
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 2011; 286:15317 - 31; http://dx.doi.org/10.1074/jbc.M110.209296; PMID: 21372138
- Hall GF, Lee S, Yao J. Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci 2002; 19:253 - 60; http://dx.doi.org/10.1385/JMN:19:3:251; PMID: 12540050
- Lee S, Kim W, Li Z, Hall GF. Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. In: Animal Models of Alzheimer's Disease. J Alz Dis 2011; In press (Hindawi ISBN: 978-953-307-797-0).
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Novak P, Prcina M, Kontsekova E. Tauons and prions: infamous cousins?. J Alzheimers Dis 2011; 26:413 - 30; PMID: 21694453
- Hall GF. What is the common link between protein aggregation and interneuronal lesion propagation in neurodegenerative disease? In: Neurodegenerative Diseases - Processes, Prevention, Protection and Monitoring. Chang R (ed), 2011; pp 1-17.
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11:909 - 13; http://dx.doi.org/10.1038/ncb1901; PMID: 19503072
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 2012; 7:e31302; http://dx.doi.org/10.1371/journal.pone.0031302; PMID: 22312444
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012; 73:685 - 97; http://dx.doi.org/10.1016/j.neuron.2011.11.033; PMID: 22365544
- Gómez-Ramos A, Díaz-Hernández M, Rubio A, Miras-Portugal MT, Avila J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci 2008; 37:673 - 81; http://dx.doi.org/10.1016/j.mcn.2007.12.010; PMID: 18272392
- Hall GF, Patuto BA. Is tau ready for admission to the prion club?. Prion 2012; 6:223 - 33; http://dx.doi.org/10.4161/pri.19912; PMID: 22561167
- Hall GF, Hasan SK. Tau secretion, tau toxicity and the biogenesis of CSF-tau – implications for how we view AD. In review
- Seppälä TT, Koivisto AM, Hartikainen P, Helisalmi S, Soininen H, Herukka S-K. Longitudinal changes of CSF biomarkers in Alzheimer’s disease. J Alzheimers Dis 2011; 25:583 - 94; PMID: 21460434
- Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem 1997; 68:430 - 3; http://dx.doi.org/10.1046/j.1471-4159.1997.68010430.x; PMID: 8978756
- Sjögren M, Rosengren L, Minthon L, Davidsson P, Blennow K, Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology 2000; 54:1960 - 4; http://dx.doi.org/10.1212/WNL.54.10.1960; PMID: 10822437
- van Harten AC, Kester MI, Visser PJ, Blankenstein MA, Pijnenburg YA, van der Flier WM, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med 2011; 49:353 - 66; http://dx.doi.org/10.1515/cclm.2011.086; PMID: 21342021
- Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 2011; 31:13110 - 7; http://dx.doi.org/10.1523/JNEUROSCI.2569-11.2011; PMID: 21917794
- Barten DM, Cadelina GW, Hoque N, DeCarr LB, Guss VL, Yang L, et al. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimers Dis 2011; 24:Suppl 2 127 - 41; PMID: 21422517
- Arai H, Terajima M, Miura M, Higuchi S, Muramatsu T, Machida N, et al. Tau in cerebrospinal fluid: a potential diagnostic marker in Alzheimer’s disease. Ann Neurol 1995; 38:649 - 52; http://dx.doi.org/10.1002/ana.410380414; PMID: 7574462
- Tato RE, Frank A, Hernanz A. Tau protein concentrations in cerebrospinal fluid of patients with dementia of the Alzheimer type. J Neurol Neurosurg Psychiatry 1995; 59:280 - 3; http://dx.doi.org/10.1136/jnnp.59.3.280; PMID: 7545739
- Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res 1989; 477:90 - 9; http://dx.doi.org/10.1016/0006-8993(89)91396-6; PMID: 2495152
- Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 1994; 87:554 - 67; http://dx.doi.org/10.1007/BF00293315; PMID: 7522386
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 1999; 58:188 - 97; http://dx.doi.org/10.1097/00005072-199902000-00008; PMID: 10029101
- Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, Vanmechelen E, et al. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett 2001; 297:187 - 90; http://dx.doi.org/10.1016/S0304-3940(00)01697-9; PMID: 11137759
- Palmio J, Suhonen J, Keränen T, Hulkkonen J, Peltola J, Pirttilä T. Cerebrospinal fluid tau as a marker of neuronal damage after epileptic seizure. Seizure 2009; 18:474 - 7; http://dx.doi.org/10.1016/j.seizure.2009.04.006; PMID: 19428269
- Siman R, McIntosh TK, Soltesz KM, Chen Z, Neumar RW, Roberts VL. Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol Dis 2004; 16:311 - 20; http://dx.doi.org/10.1016/j.nbd.2004.03.016; PMID: 15193288
- Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic 2005; 6:607 - 14; http://dx.doi.org/10.1111/j.1600-0854.2005.00302.x; PMID: 15998317
- Chen C, Shi Q, Zhang BY, Wang GR, Zhou W, Gao C, et al. The prepared tau exon-specific antibodies revealed distinct profiles of tau in CSF of the patients with Creutzfeldt-Jakob disease. PLoS One 2010; 5:e11886; http://dx.doi.org/10.1371/journal.pone.0011886; PMID: 20686702
- Tosun D, Schuff N, Truran-Sacrey D, Shaw LM, Trojanowski JQ, Aisen P, et al, Alzheimer’s Disease Neuroimaging Initiative. Relations between brain tissue loss, CSF biomarkers, and the ApoE genetic profile: a longitudinal MRI study. Neurobiol Aging 2010; 31:1340 - 54; http://dx.doi.org/10.1016/j.neurobiolaging.2010.04.030; PMID: 20570401
- Solé-Padullés C, Lladó A, Bartrés-Faz D, Fortea J, Sánchez-Valle R, Bosch B, et al. Association between cerebrospinal fluid tau and brain atrophy is not related to clinical severity in the Alzheimer’s disease continuum. Psychiatry Res 2011; 192:140 - 6; http://dx.doi.org/10.1016/j.pscychresns.2010.12.001; PMID: 21546220
- Snow J. On the Mode of Communication of Cholera. 2nd ed, London, J. Churchill, 1855, pp 162.