Abstract
Lowe syndrome (LS) is a lethal X-linked genetic disease caused by functional deficiencies of the phosphatidlyinositol 5-phosphatase, Ocrl1. In the past four years, our lab described the first Ocrl1-specific cellular phenotypes using dermal fibroblasts from LS patients. These phenotypes, validated in an ocrl1-morphant zebrafish model, included membrane remodeling (cell migration/spreading, fluid-phase uptake) defects and primary cilia assembly abnormalities. On one hand, our findings unraveled cellular phenotypes likely to be involved in the observed developmental defects; on the other hand, these discoveries established LS as a ciliopathy-associated disease. This article discusses the possible mechanisms by which loss of Ocrl1 function may affect RhoGTPase signaling pathways leading to actin cytoskeleton rearrangements that underlie the observed cellular phenotypes.
The Oculo-Cerebro-Renal syndrome of Lowe (OCRL) or Lowe Syndrome (LS) is a recessive X-linked disease characterized by congenital cataracts, mental retardation and renal dysfunction. Unfortunately, affected children die at an early age, most commonly of renal failure.
This devastating genetic disorder was first described by Drs. Lowe, Terrey, and MacLachlanCitation1 about 60 years ago and its association with mutations in the OCRL1 gene was established in the early nineties by a team led by Dr. Robert Nussbaum.Citation2 The OCRL1 gene product is the Inositol 5-phosphatase Ocrl1 (EC 3.1.3.36) that recognizes and hydrolyzes PhosphatidylInositol (PtdIns) 5-phosphates, including the plasma membrane-enriched lipid - PtdIns(4,5) bis-phosphate (PtdIns(4,5)P2). Despite this knowledge, the actual mechanism by which deficiencies in Ocrl1 function leads to this debilitating disease still remains unclear.
In that respect, we recently established that LS patient cells display defects in the assembly of primary cilia;Citation3 and had previously shown that the same cells exhibit severe abnormalities in Rac1-mediated, membrane remodeling processes.Citation4-Citation6 These findings represent significant advances in our understanding of the physiological roles of Ocrl1, but they also pose new questions. For example, is Ocrl1 a protein with two distinct functions or are these phenotypes different manifestations of the same basic functional abnormality? Second, how do these newly observed Ocrl1-specific cellular phenotypes contribute toward better understanding of LS?
Membrane-Remodelling Phenotypes in LS Cells
We identified abnormalities in cell migration, cell spreading and fluid-phase uptake in primary cultured skin fibroblasts from two unrelated LS patients. The cell migration defect was detected by two independent assays: transwell-based and “wound”-healing.Citation4 Notably, Ocrl1 phosphatase function and its interaction with the endocytic machinery (clathrin and the endocytic adaptor AP2) were both required for its function in cell migration.Citation4 LS cells also failed to spread isotropically when seeded on fibronectin-coated surfaces.Citation4 Consequently, the size distribution of LS patient fibroblasts was significantly skewed toward cells of smaller area as compared with fibroblasts from normal individuals.Citation4 In addition, the LS cell population was enriched in irregularly shaped cells (), likely representing early spreading intermediates.Citation4
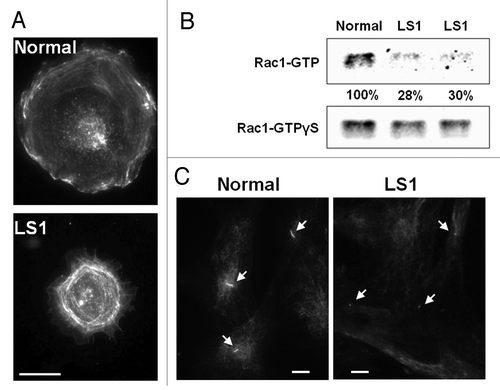
Figure 1. Cellular phenotypes of Lowe syndrome patient cells. A. Cell spreading abnormalities. Normal and LS cells were resuspended and seeded on fibronectin-coated surfaces. After 30 min, the cells were fixed with 4% formaldehyde, stained with rhodamine–phalloidin and imaged. Scale bar: 20 microns. B.Rac1 activation deficiencies. Lysates from Normal and LS patient (LS1 and LS2) fibroblasts were incubated with GST-PAK1 (CRIB) beads that bind (activated) Rac1-GTP. Bound Rac1-GTP was detected by western blotting using an anti-Rac1 antibody. Results were analyzed by band densitometry. GTPγS -loaded lysates contained equivalent amounts of Rac1 in all samples. Levels of Rac1-GTP (normalized relative to corresponding GTPγS -loaded Rac1 signal) were calculated as a fraction of Normal cell values. C.Primary cilia assembly defects. Dermal fibroblasts from a normal individual and a LS patient (LS1) were grown on coverslips and induced to form primary cilia by serum-starvation. Following fixation with 4% formaldehyde the cells were immunostained using an anti-acetylated tubulin specific antibody. Arrows point to primary cilia. Scale bar: 20 microns.
The developing embryo relies on perfect timing and synchronization of the above mentioned processes, e.g., during organogenesis. Therefore, we speculated that the LS cellular phenotypes would have important developmental consequences. Indeed, eye abnormalities observed in LS patients have been suggested to be linked to migration defects of lens cells.Citation7 Further, a zebrafish model of LS confirmed the existence of defects in cell migration that affected melanocytes as well as neural crest cells (NCC).Citation3 Further, this animal model also displayed developmental defects, likely due to NCC migration defects, such as facial dysmorphology consisting of short and malformed palatoquadrate and Meckel’s cartilage, components of the mandibular arch, and missing or smaller pharyngeal arches.Citation3
What is the mechanism by which Ocrl1-deficiency affects cell migration/spreading?
Dynamic actin cytoskeletal rearrangements are required for membrane remodeling processes, particularly for cell migration and spreading. In fact, previous studies have reported deficiencies in actin organization in LS cells, namely decreased stress fiber staining, punctuate actin structuresCitation8 and abundant actin “comets.”Citation9 Interestingly, Faucherre et al.Citation10 demonstrated that Ocrl1 directly binds to the RhoGTPase Rac1, a crucial regulator of actin dynamics.Citation11 In fact, our lab found that the level of activated Rac1 is significantly decreased in LS fibroblasts ().Citation6 Importantly, Rac1 activation is required for stabilizing lamellopodium extension during cell spreadingCitation12 and to sustain velocity and directional persistence during migration.Citation13,Citation14 Using time-lapse videomicroscopy, we observed that LS cells cannot sustain cell spreading for long periods of time and eventually retract.Citation5,Citation6 Patient cells also display a decrease in velocity and directional persistence during migration (unpublished data).Citation5,Citation6 In addition, Lasne et al. suggested aberrant signaling by the RhoGTPase RhoA in platelets from LS patient cells.Citation15 Taken together, evidence collected by our lab and others suggests that LS patients show RhoGTPase signaling abnormalities that lead to membrane remodeling abnormalities.
What is the mechanism leading to RhoGTPase signaling abnormalities in LS patients?
We speculate that functional deficiencies in Ocrl1 may lead to abnormal RhoGTPase signaling by at least two non-exclusive mechanisms:
i) Defects in the activation of Rac1 in endomembranes: It has been demonstrated that Rac1 is activated in endosomes by its GEF Tiam1, following which it is recycled to the leading edge of migratory cells.Citation16 Since Ocrl1 is involved in endosomal trafficking,Citation17,Citation18 it is possible that subsequent to Rac1 binding, the Lowe syndrome protein facilitates the activation of the RhoGTPase and perhaps its re-routing to the leading edge.
ii) PtdIns imbalance: It has been demonstrated that the loss of Ocrl1 leads to elevated levels of PtdIns(4,5)P2.Citation19 Indeed, Ueno and coworkers observed that high levels of PtdIns(4,5)P2, in the presence of PI(4)P (product of Ocrl1 phosphatase activity) could lead to membrane ruffles through Rac1 activation, whereas high levels of PtdIns(4,5)P2, by consumption of PI(4)P lead to endosomal comet tails via RhoA activation.Citation20 Since RhoKinase inhibitor and dominant negative Rac1 had opposing effects on the abundance of comet tails, a reciprocal antagonism between the Rac/RhoA GTPases has been suggested.Citation20 Further, the authors proposed that defective 5-phosphatase activity toward PtdIns(4,5)P2 in LS fibroblasts followed by abnormal Rac/RhoA activation could be responsible for the inability of LS fibroblasts to migrate properly.Citation20
Therefore, probing the mechanisms underlying decreased levels of GTP-bound Rac1 and the related cellular phenotypes in LS patients will be an interesting subject for future investigations.
Primary Cilia Assembly Abnormalities in LS Patient Cells
We recently discovered another novel cellular phenotype associated with LS: deficiencies in primary cilia (PC) assembly.Citation3 The PC is an axoneme-based sensing organelle which plays a key role during development.Citation21 Importantly, deficiencies in PC assembly are the recognized cause of a broad, heterogeneous group of pathologies collectively known as ciliopathies.Citation22 Interestingly, similar to LS, ciliopathy patients also display symptoms affecting the brain, eyes and kidneys.Citation22,Citation23
Specifically, we found that upon serum-starvation, fewer LS patient fibroblasts were able to assemble PC as compared with controls. Moreover, the PC displayed by the LS cells were shorter and rudimentary as compared with their normal counterparts ().Citation3 Mechanistically, our studies indicated that Ocrl1 participates in vesicle trafficking to the PC and that its interaction with its endosomal binding partners Appl1 and IPIP27/Ses plays a crucial role in this novel Ocrl1 function.Citation3
It should be highlighted that zebrafish ocrl1-morphants also showed gross cilia disorganization and shorter cilia in their pronephros as compared with their normal counterparts.Citation3 Our zebrafish model also showed developmental defects reminiscent of those seen in ciliopathies, such as NCC patterning defects and facial dysmorphism.
Our findings indicate that LS shares characteristics with ciliopathies. In fact, this conclusion changes the status of the disease from being an isolated syndrome to a ciliopathy-associated pathology. Therefore, we anticipate that advances made in LS research will have an impact in the field of ciliopathies and vice versa.
Importantly, our studies were followed by two other reports that confirmed the role of Ocrl1 in ciliary biogenesis.Citation24,Citation25
LS: Ciliopathy-Associated or Membrane-Remodeling Pathology? Connecting the Dots
As discussed above and elsewhere,Citation3,Citation24-Citation26 LS displays similarities with ciliopathies. In fact, defects in cell migration and spreadingCitation22,Citation23 as well as RhoGTPase signaling abnormalitiesCitation27 have also been observed in ciliopathies. However, is the PC assembly abnormality linked to membrane remodeling defects in LS?
Rho GTPase activation has been shown to mediate apical actin enrichment required for ciliogenesis,Citation28,Citation29 but it is also known that signaling events initiated at the PC lead to Rac1/RhoA activation.Citation27 Therefore, whether RhoGTPase signaling defects are the cause or consequence of PC defects should be the subject of further investigation.
It should be also noted that while membrane remodeling is induced upon cell stimulation with growth factors, PC stabilization requires serum-“starvation” conditions.Citation3,Citation4 Therefore, Ocrl1 participation in one and the other function occur under very different conditions; suggesting that these functions are independent of each other.
Our data also indicated a differential requirement of the molecular interactions of Ocrl1 for sustaining membrane remodeling vs. PC assembly. Most noticeably, Ocrl1 binding to clathrin and AP-2 was required for its ability to support cell migration/spreading but was dispensable for PC assembly.Citation3,Citation4 Another noteworthy observation was that Inpp5b, the autosomal paralog of Ocrl1, was unable to uphold any of the membrane remodeling processes, but showed partial competence in sustaining PC assembly.Citation3,Citation4
Therefore, we hypothesize that Ocrl1 relies on different protein-protein interactions to support different cellular functions (membrane remodeling/Rac1 activation vs. vesicle trafficking to the PC). Nevertheless, we do not discard the idea that these different Ocrl1 functions may co-operate with each other to support specific processes. For example, the PC plays a role in establishing the direction of migration;Citation30 therefore, it is expected that Ocrl1 deficiency may affect cell crawling via different mechanisms (membrane remodeling and functional PC signaling). Perhaps, depending on the cell type or the presence of specific stimuli during organogenesis, Ocrl1 deficiency may have differential impacts during embryo development.
How do these findings affect our ideas about and approaches toward LS?
Though LS and other ciliopathies affect common organs and share various phenotypes, they do differ in the specifics of the symptoms suffered by patients (e.g., cataracts vs. retinitis pigmentosa; renal tubulopathy vs. renal cysts). These differences remain to be explained. Nevertheless, something we have already learnt from the ciliopathy field is that alterations in the functionality of a single organelle, pathway or gene can lead to a plethora of different symptoms.Citation22,Citation23 For example, mutations in the CEP290 gene can lead not only to Bardet-Biedl syndrome, but also to Meckel syndrome, nephronophthisis and Joubert syndrome. It should be noted that each of these pathologies has its own set of clinical manifestations. Similarly, relevant to this discussion is the OCRL1 gene itself, mutations in which causes both Lowe syndrome and Dent-2 disease.Citation31 Among several possible reasons for this phenotypic variability, mutation-specific effects on protein-protein interactions, presence of modifier loci, post-translational modifications and epigenetics are prime suspects.
We expect that these discoveries will set in motion synergistic interactions between investigators working on Lowe syndrome and ciliopathy-associated disorders. Indeed, we hope that breakthroughs in terms of mechanistic details or novel therapeutic strategies, arising in the field of ciliopathies, could be immediately capitalized by LS researchers and vice versa.
Acknowledgments
We thank Arpita Sen (Purdue University) for critical reading of the manuscript and various colleagues for insightful discussions and suggestions. The Aguilar lab research on LS is supported by grants from the Lowe Syndrome Trust (UK). K.M. is supported by a fellowship from the Cancer Prevention Interdisciplinary Program at Purdue University, NIH grant R25CA128770.
Note added in proof
While this manuscript was under formatting, an article describing diminished Rac1 activation (and high levels of RhoAGTP) in LS cells was published (van Rahden VA, Brand K, Najm J, Heeren J, Pfeffer SR, Braulke T, Kutsche K.Hum Mol Genet. 2012. doi: 10.1093/hmg/dds343; PMID: 22907655). This work confirms some of our findings (particularly deficiencies in Rac1 activation upon Ocrl deficiency) and also indicates abnormal Cofilin function downstream of Rac1. In addition, this paper presents a very rigorous and detailed study of Ocrl1 function in vesicle trafficking.
References
- Lowe CU, Terrey M, Maclachlan EA. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation - a clinical entity. Am J Dis Child 1952; 83
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, et al. The Lowe Oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 1992; 358
- Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I, et al. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet 2012; 21:1835 - 47; http://dx.doi.org/10.1093/hmg/ddr615; PMID: 22228094
- Coon BG, Mukherjee D, Hanna CB, Riese DJ 2nd, Lowe M, Aguilar RC. Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5b phosphatase. Hum Mol Genet 2009; 18:4478 - 91; http://dx.doi.org/10.1093/hmg/ddp407; PMID: 19700499
- Coon BG, Mukherjee D, Hanna CB, Reise DJ 2nd, Lowe M, Aguilar RC. Lowe Syndrome patient fibroblasts display OCRL1-specific cell migration defects that cannot be rescued by the homologous INPP5b phosphatase. Mol Biol Cell 2009; 20:81 Abstract# 917
- Aguilar RC. OCRL1 deficiency leads to cell migration defects in Lowe Syndrome patient cells. Mol Biol Cell 2009; 20:12
- Loi M. Lowe syndrome. Orph J Rare Diseas 2006; 1.
- Suchy SF, Nussbaum RL. The deficiency of PIP2 5-phosphatase in Lowe syndrome affects actin polymerization. Am J Hum Genet 2002; 71:1420 - 7; http://dx.doi.org/10.1086/344517; PMID: 12428211
- Allen PG. Actin filament uncapping localizes to ruffling lamellae and rocketing vesicles. Nat Cell Biol 2003; 5:972 - 9; http://dx.doi.org/10.1038/ncb1059; PMID: 14557819
- Faucherre A, Desbois P, Satre V, Lunardi J, Dorseuil O, Gacon G. Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. Hum Mol Genet 2003; 12:2449 - 56; http://dx.doi.org/10.1093/hmg/ddg250; PMID: 12915445
- Hall A. Rho GTPases and the actin cytoskeleton. Science 1998; 279:509 - 14; http://dx.doi.org/10.1126/science.279.5350.509; PMID: 9438836
- Yamazaki D, Fujiwara T, Suetsugu S, Takenawa T. A novel function of WAVE in lamellipodia: WAVE1 is required for stabilization of lamellipodial protrusions during cell spreading. Genes Cells 2005; 10:381 - 92; http://dx.doi.org/10.1111/j.1365-2443.2005.00845.x; PMID: 15836768
- Pankov R, Endo Y, Even-Ram S, Araki M, Clark K, Cukierman E, et al. A Rac switch regulates random versus directionally persistent cell migration. J Cell Biol 2005; 170:793 - 802; http://dx.doi.org/10.1083/jcb.200503152; PMID: 16129786
- Chae YC, Kim JH, Kim KL, Kim HW, Lee HY, Heo WD, et al. Phospholipase D activity regulates integrin-mediated cell spreading and migration by inducing GTP-Rac translocation to the plasma membrane. Mol Biol Cell 2008; 19:3111 - 23; http://dx.doi.org/10.1091/mbc.E07-04-0337; PMID: 18480413
- Lasne D, Baujat G, Mirault T, Lunardi J, Grelac F, Egot M, et al. Bleeding disorders in Lowe syndrome patients: evidence for a link between OCRL mutations and primary haemostasis disorders. Br J Haematol 2010; 150:685 - 8; http://dx.doi.org/10.1111/j.1365-2141.2010.08304.x; PMID: 20629659
- Palamidessi A, Frittoli E, Garré M, Faretta M, Mione M, Testa I, et al. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell 2008; 134:135 - 47; http://dx.doi.org/10.1016/j.cell.2008.05.034; PMID: 18614017
- Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, et al. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell 2007; 13:377 - 90; http://dx.doi.org/10.1016/j.devcel.2007.08.004; PMID: 17765681
- Cui S, Guerriero CJ, Szalinski CM, Kinlough CL, Hughey RP, Weisz OA. OCRL1 function in renal epithelial membrane traffic. Am J Physiol Renal Physiol 2010; 298:F335 - 45; http://dx.doi.org/10.1152/ajprenal.00453.2009; PMID: 19940034
- Zhang XL, Hartz PA, Philip E, Racusen LC, Majerus PW. Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J Biol Chem 1998; 273:1574 - 82; http://dx.doi.org/10.1074/jbc.273.3.1574; PMID: 9430698
- Ueno T, Falkenburger BH, Pohlmeyer C, Inoue T. Triggering actin comets versus membrane ruffles: distinctive effects of phosphoinositides on actin reorganization. Sci Signal 2011; 4:pt4; PMID: 21693763
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 2010; 11:331 - 44; http://dx.doi.org/10.1038/nrg2774; PMID: 20395968
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364:1533 - 43; http://dx.doi.org/10.1056/NEJMra1010172; PMID: 21506742
- Schurman SJ, Scheinman SJ. Inherited cerebrorenal syndromes. Nat Rev Neurol 2009; 5:529 - 38; PMID: 19794512
- Luo N, West CC, Murga-Zamalloa CA, Sun L, Anderson RM, Wells CD, et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet 2012; 21:3333 - 44; http://dx.doi.org/10.1093/hmg/dds163; PMID: 22543976
- Rbaibi Y, Cui S, Mo D, Carattino M, Rohatgi R, Satlin LM, et al. OCRL1 modulates cilia length in renal epithelial cells. Traffic 2012; 13:1295 - 305; http://dx.doi.org/10.1111/j.1600-0854.2012.01387.x; PMID: 22680056
- Conduit SE, Dyson JM, Mitchell CA. Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Lett 2012; 586:2846 - 57; http://dx.doi.org/10.1016/j.febslet.2012.07.037; PMID: 22828281
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet 2010; 42:619 - 25; http://dx.doi.org/10.1038/ng.594; PMID: 20512146
- Pan J, You Y, Huang T, Brody SL. RhoA-mediated apical actin enrichment is required for ciliogenesis and promoted by Foxj1. J Cell Sci 2007; 120:1868 - 76; PMID: 17488776
- Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet 2008; 40:871 - 9; http://dx.doi.org/10.1038/ng.104; PMID: 18552847
- Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR, et al. Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell Physiol Biochem 2010; 25:279 - 92; http://dx.doi.org/10.1159/000276562; PMID: 20110689
- Shrimpton AE, Hoopes RR Jr., Knohl SJ, Hueber P, Reed AAC, Christie PT, et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron, Physiol 2009; 112:27 - 36; http://dx.doi.org/10.1159/000213506