
Abstract
Neuronal abnormalities in neurodegenerative disorders such as Huntington disease, Alzheimer disease or Parkinson disease have been the primary focus of decades of research. However, increasing evidences indicate that glial cells and more specifically astrocytes could be as important players as their big brother. It is now particularly evident in Huntington disease where astrocytal potassium channels have emerged as a likely key factor in the pathogenesis of the disease.
Huntington disease (HD) is an inherited neurodegenerative disorder, caused by an autosomal dominant mutation in the Huntingtin gene (HTT).Citation1 In 1872, Gorge Huntington first described the medical history of several generations of a family experiencing similar symptoms, suggesting that these symptoms must be linked.Citation2 The disease was later named after him and is characterized by alterations in personality, cognition, and motor control, associated with neuronal dysfunction and atrophy of the striatum, the substantia nigra, the CA1 region of the hippocampus, and in a lesser extent other brain regions.Citation3 HD is much more common in Europe, North America, and Australia than it is in Asia, with a prevalence of 5–7 and 0.4 affected individuals per 100.000, respectively. This difference is mainly explained by the existence of various huntingtin gene haplotypes.Citation3,Citation4 The cause of HD is a well-known expansion of CAG trinucleotides located at the N-terminal region of the huntingtin protein (HTT).Citation5 Mutated huntingtin (mHTT) tends to aggregate and form intracellular accumulations, a common cellular phenotype also commonly observed with other neurodegenerative diseases.Citation6,Citation7 The physiological role of HTT as well as the pathological mechanism of the disease is largely unknown. However, there are a few known interaction partners that link HTT to vesicular transport, cytoskeletal organization, presynaptic signaling, and anti-apoptotic factors.Citation8 The initial observation that mHTT accumulates in striatal astrocytesCitation9,Citation10 and the fact that ion channels have been implicated in neurodegenerative diseases earlierCitation11 lead Tong et al.Citation12 to investigate the functional implication of glial cells in the pathophysiology of HD. Using mouse models of HD, the authors demonstrate that striatal astrocytes containing mHTT present altered Kir4.1 channel activity and aberrant extracellular potassium homeostasis rendering striatal neurons hyperexcitable, which possibly account for HD motor symptoms.
Intracellular inclusions of mHTT were found in striatal astrocytes of R6/2 mice at a symptomatic stage of HD, before any signs of astrogliosis. Astrogliosis is defined as an increase in astrocyte population due to destruction of nearby neurons, also often observed in brain trauma, stroke, ischemia, and neurodegenerative diseases. Indeed, while old R6/2 mice (P104-P110) with established neurodegeneration and striatal atrophy present an increase in Glial Fribrillary Acidic Protein signal (GFAP, an astrocyte marker), GFAP reactivity remained unaltered in symptomatic R6/2 mice (P60–80) indicating that HD symptoms start before any astrogliosis. In addition, pSTAT3 (Signal Transducer and Activator of Transcription 3), a transcription regulator usually increased during astrogliosis was found stable in symptomatic R6/2 mice compare with wild type animals.
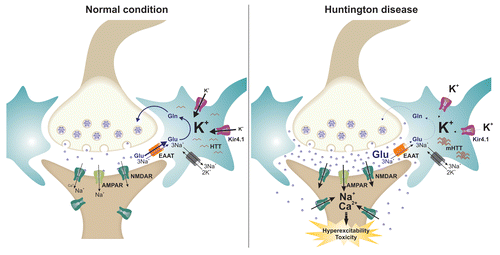
Using another mouse model of HD, the authors found that striatal astrocytes from HDR6/2 mice show altered electrical properties including a decreased membrane conductance and depolarized resting membrane potential. In addition, the inward-rectifying Kir4.1 potassium (K+) channel activity was found significantly reduced. Kir4.1 channel is one of the predominant astrocytal K+ channelsCitation13-Citation15 that contribute to establish the resting membrane potential of the cells by passing more easily K+ ions inside the cell than outside, and also responsible for buffering extracellular K+ in the brain. Mutations in Kir4.1 channel are associated with some forms of epilepsy and ataxia.Citation16 Patch-clamp recordings on astrocytes isolated from R6/2 and Q147 HD mouse indicate that the Ba2+ sensitive K+ current, i.e., the K+ current conducted by Kir channels, is significantly reduced at the symptomatic stage but not at a pre-symptomatic time. Reduced Kir currents are most likely the result of either an alteration in Kir channels expression at the plasma membrane or from a direct alteration of channel activity since no change in Kir4.1 mRNA was noticed in R6/2 striatal astrocytes. Consistent with an altered Kir4.1 activity, extracellular potassium concentration was found increased from 1.5 mM to 3 mM, and similar increase in K+ concentration applied on striatal slice from wild type animals was sufficient to make neurons hyperexcitable. Finally, viral delivery of Kir4.1 channel into R6/2 mouse striatum was found sufficient to rescue astrocyte electrical properties and normalize extracellular K+ level, while only some of the motor symptoms were improved in mice, indicating that Kir4.1 channels contribute to HD symptoms but most likely are not sufficient to fully explain the pathogenesis of the disease. Hence, a decreased expression of the glutamate transporter EAAT (Excitatory Amino-Acid Transporter), which play a critical role in removing glutamate from the synaptic cleft and thus terminating synaptic transmissionCitation17 was also observed in R6/2 striatum.
While implication of Kir4.1 channel is unambiguous, the cellular mechanisms by which alteration in channel activity contributes to HD symptoms remain intriguing. It is possible that Kir4.1-dependent alteration of neuronal excitability could involve glutamate signaling as it has already been proposed.Citation9,Citation10 Indeed, while activity of the EAAT relies on extracellular Na+, it also strongly depends on transmembrane K+/H+ concentration gradient.Citation18 Given the decreased activity of Kir4.1 channel and subsequent increase in extracellular K+ concentration in HD mouse models, it is reasonable to think that activity of the glutamate transporter could be altered in those animals. Decrease in EAAT expression, combined with a decreased activity of the transporter could in turn let the extracellular glutamate building up and cause neuronal hyperexcitability and in the long-term cellular toxicity, possibly by acting on extrasynaptic NMDA receptors that have been particularly implicated in glutamate-associated toxicityCitation19 (see also ). Further investigations to assess extracellular glutamate homeostasis in those HD animal models will certainly provide important insights into the cellular and molecular mechanisms by which astrocytal Kir4.1 channels contribute to HD symptoms. Moreover, considering that an alteration in Kir4.1 channel expression and altered K+ homeostasis was also reported in animal mouse models of Alzheimer disease,Citation20 Kir4.1 channel appears as a common underlying mechanism of those neurological disorders and possibly opens interesting new therapeutic avenues.
Figure 1. A putative model linking astrocytal Kir4.1 channel and glutamate homeostasis in Huntington disease. In normal condition, glutamate released in the synaptic cleft is rapidly removed by astroctal EAAT transporters that serve o terminate the excitatory signal and preventing neuronal excitotoxicity. Under pathological condition such as Huntington disease, decreased Kir4.1 channel activity leads to aberrant K+ homeostasis and transmembrane K+ gradient disturbing EAAT activity and astrocytal glutamate uptake. Accumulation of glutamate in the synaptic cleft causes in turn neuronal hyperexcitability and in the long-term cellular toxicity, possibly by activation of extrasynaptic NMDA receptors.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
J.P. is supported by a postdoctoral fellowship from the Institute of Organic Chemistry and Biochemistry and the Academy of Sciences of the Czech Republic.
References
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014; 10:204 - 16; http://dx.doi.org/10.1038/nrneurol.2014.24; PMID: 24614516
- G H. On Chorea. Med Surg Rep 1872; 26:317 - 21
- Walker FO. Huntington’s Disease. Semin Neurol 2007; 27:143 - 50; http://dx.doi.org/10.1055/s-2007-971176; PMID: 17390259
- Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 2012; 27:1083 - 91; http://dx.doi.org/10.1002/mds.25075; PMID: 22692795
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2006; 2:679 - 89; http://dx.doi.org/10.1038/ncpneuro0355; PMID: 17117171
- Proft J, Weiss N. A protective mutation against Alzheimer disease?. Commun Integr Biol 2012; 5:301 - 3; http://dx.doi.org/10.4161/cib.21799; PMID: 23060947
- Proft J, Weiss N. Jekyll and Hide: The two faces of amyloid β. Commun Integr Biol 2012; 5:405 - 7; http://dx.doi.org/10.4161/cib.22571; PMID: 23181153
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell 2004; 15:853 - 65; http://dx.doi.org/10.1016/j.molcel.2004.09.016; PMID: 15383276
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 2005; 171:1001 - 12; http://dx.doi.org/10.1083/jcb.200508072; PMID: 16365166
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet 2010; 19:3053 - 67; http://dx.doi.org/10.1093/hmg/ddq212; PMID: 20494921
- Proft J, Weiss N. T-type Ca(2+) channels: New players in the aging brain. Commun Integr Biol 2014; 7:e28424; http://dx.doi.org/10.4161/cib.28424; PMID: 24748914
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 2014; 17:694 - 703; http://dx.doi.org/10.1038/nn.3691; PMID: 24686787
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol 2001; 281:C922 - 31; PMID: 11502569
- Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, Gallo V, Wilkin GP. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia 2000; 30:362 - 72; http://dx.doi.org/10.1002/(SICI)1098-1136(200006)30:4<362::AID-GLIA50>3.0.CO;2-4; PMID: 10797616
- Barres BA, Koroshetz WJ, Chun LL, Corey DP. Ion channel expression by white matter glia: the type-1 astrocyte. Neuron 1990; 5:527 - 44; http://dx.doi.org/10.1016/0896-6273(90)90091-S; PMID: 1698397
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 2009; 360:1960 - 70; http://dx.doi.org/10.1056/NEJMoa0810276; PMID: 19420365
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature 1996; 383:634 - 7; http://dx.doi.org/10.1038/383634a0; PMID: 8857541
- Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65:1 - 105; http://dx.doi.org/10.1016/S0301-0082(00)00067-8; PMID: 11369436
- Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014; 82:279 - 93; http://dx.doi.org/10.1016/j.neuron.2014.03.030; PMID: 24742457
- Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009; 159:1055 - 69; http://dx.doi.org/10.1016/j.neuroscience.2009.01.023; PMID: 19356689