Abstract
Abstract Williams syndrome (a.k.a. Williams-Beuren Syndrome) is a multisystem disorder caused by the hemizygous deletion of a 1.6-Mb region at 7q11.23 encompassing about 26 genes, including that encoding LIM kinase 1 (LIMK1). Individuals with Williams Syndrome manifest hyperacusis and progressive hearing loss, and hyperacusis early onset suggests that it could be associated with one of the deleted genes. Based on our results about the critical role of LIM kinases in the regulation of the motile response of cochlear outer hair cells and cochlear amplification, we propose here that a reduced expression of LIMK1 in outer hair cells would be the major underlying cause of the hyperacusis and progressive hearing loss observed in patients with Williams Syndrome. Moreover, we propose a novel model of gain-control for cochlear amplification based on the LIMK-mediated regulation of slow motility of cochlear outer hair cells.
Williams syndrome (WS) is a neurodevelopmental disorder caused by a hemizygous microdeletion of approximately 1.6 Mb on the long arm of chromosome 7 (7q11.23) containing about 26 genes, including LIMK1 and elastin.Citation1–Citation5 In addition to cardiovascular anomalies, moderate mental retardation and severe impairments in visuospatial processing, WS patients show oversensitivity to sounds, including hyperacusis, phonophobia and auditory fascination.Citation6 The early onset and pervasiveness of hyperacusis in individuals with WS suggests that it could be associated with one of the deleted genes.Citation7
Hyperacusis is defined as an over-sensitivity to sound associated with an excessive auditory gain because of a dysfunction in the mechanism of cochlear amplification.Citation8,Citation9 Excessive auditory gain naturally implies an intact cochlear amplifier. This points at a failure in controlling the gain of the amplification mechanism, rather than a failure in the mechanism itself, as the culprit for hyperacusis in WS subjects. Audiometric studies, DPOAE and TEOAE recordings strongly suggest that the WS subjects have OHC dysfunction.Citation5,Citation10–Citation12 In addition, studies involving OHC modulation by the ipsilateral medial olivocochlear (MOC) system showed that the mechanism affected in WS subjects is a target of acetylcholine (ACh),Citation8 the major neurotransmitter released by efferent terminals at the base of OHCs.Citation13 In normal individuals, electrical stimulation of MOC fibers reduces cochlear amplification by some 20 dB, a central result to the widely accepted hypothesis of efferent-mediated protection of auditory function from noise trauma.Citation13,Citation14
The possible origin of the dysfunction in cochlear amplification causing hyperacusis in WS subjects is still a matter of debate.Citation5,Citation6,Citation10,Citation15 Elastin insufficiency was considered a possible cause based on early reports describing the destruction of hair bundle tip-links with the enzyme elastase.Citation16–Citation18 It was hypothesized that tip-links could be composed by elastin, and elastin deficiency could lead to some kind of desynchronized movement of the stereocilia and a combination of hearing loss and acoustic nerve dysfunction responsible for an altered perception of loudness in the afferent auditory system.Citation7 More recent studies indicating that tip-links might actually be composed by cadherin-23 and proto-cadherin 15 make this hypothesis unlikely.Citation19–Citation22
An alternative gene that might be responsible for the auditory phenotype in WS is LIMK1, which encodes for a serine/threonine kinase that regulates actin reorganization.Citation23 LIMK1 gene localizes in the middle of the region deleted in individuals with WS, and LIMK1 knockout mice subjected to a fear-conditioning test showed significantly longer and more constant freezing than wild-type mice when exposed to certain sounds.Citation24 However, it remains unclear if that aggravated responses in KO mice were specific to auditory stimuli.Citation6 The gene encoding the only other known member of the LIMK family, LIMK2, localizes in human chromosome 22 (22q12) and it has not been associated with WS. To our knowledge, our recent publication is the first report linking LIMK to OHC motility and cochlear amplification, and the first suggesting that “any disruption in the signaling pathways involving these molecules could result in extreme physiological responses such as hyperacusis or deafness.”Citation25 Thus, based on our original results, we are proposing here a deficiency in LIMK1 as the potential cause of WS-associated hyperacusis.
We found that activation or inhibition of LIMK-mediated pathways regulate cochlear amplification by increasing or decreasing, respectively, both electromotile amplitude and total length of cochlear OHCs without any effect on the performance of the plasma membrane-embedded motor (prestin) proteins.Citation25 Since LIMK1 is absent in OHC's stereocilia, we consider unlikely any significant effect on any active process in the stereocilia bundle putatively associated with cochlear amplification. Thus, we speculate that a deficient expression of LIMK1 would result in a simultaneous decrease of both electromotile amplitude and OHC total length. This is a very important point that reveals the fine-tuning mechanism underlying homeostatic control of cochlear amplification. Whereas OHC shortening would be associated with an increase in the gain of the cochlear amplifier (see below and ), a decrease in OHC electromotility is generally associated with gain decrease. Thus, the two OHC motile responses would be working in a dynamic equilibrium, with any change in OHC length moving the gain of the cochlear amplifier either up or down being simultaneously counteracted by a change in electromotile amplitude pushing the gain in the opposite direction.
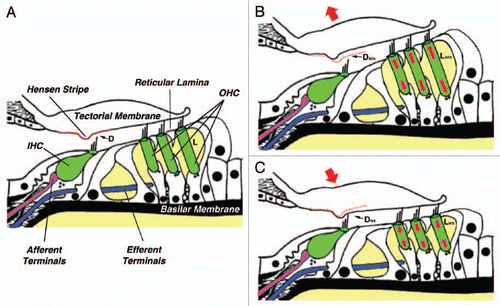
How does LIMK1 deficiency could translate into hyperacusis? We propose the following mechanism ():
In a given steady-state condition (), OHCs would have a particular length L and the distance between the lower surface of the tectorial membrane (TM) and the tip of the IHCs' hair bundle would have a particular value D. Sound stimulation would induce vibration of the basilar membrane (BM), generating a shearing movement between the TM and the reticular lamina that deflects OHCs' hair bundles.Citation26 Hair bundle's deflection would open ionic channels at the tip of the stereocilia, changing the electrical potential inside the OHCs and triggering electromotile responses that amplify 1,000-fold and more the original amplitude of BM vibration. Since the tip of OHC's tallest stereocilia are embedded in the TM, electromotility would move up and down this structure creating a flow of endolymphatic fluid in the subtectorial space sufficient to deflect IHCs' hair bundles (which are not connected to the tectorial membrane), generating electrochemical signals that would be transmitted to the brain via the afferent terminals innervating the IHCs.Citation27,Citation28
-In the event of high intensity sound stimulation the brain would protect the IHCs by activating a negative feedback mechanism, based on the release of ACh by the MOC efferent terminals innervating the OHCs, aimed at decreasing the gain of the cochlear amplifier (). ACh would increase the concentration of intracellular Ca2+, activating Ca2+-dependent, LIMK-mediated pathways targeting the OHC cytoskeleton and triggering slow motile responses leading to a significant increase in both total OHC length (LACh>L) and electromotile amplitude. The increase in total OHC length would increase the distance between the TM and the tip of IHCs' stereocilia (DACh>D), diminishing the speed of the flow of endolymph and, consequently, the deflection of IHCs' stereocilia and the gain of the cochlear amplifier.
-Genetic or physiological problems leading to total disruption or malfunctioning of the mechanism/s regulating OHC motility, such as LIMK1 deficiency in WS subjects, could be the underlying cause of hyperacusis (). Abnormal signaling could result in excessive and/or uncontrolled shortening of OHCs (LWS <L) leading to a decrease in the distance between the TM and the tip of IHCs' stereocilia (DWS<D). This change in the geometry of the subtectorial space would increase the speed of the flow of endolymph and consequently, the deflection of IHCs' stereocilia and the gain of the cochlear amplifier. In addition, the displacement of the TM would close the gap between the Hensen stripe and the hair bundle of the IHCs, increasing the risk of mechanical damage of the IHC's hair bundle. This damage could be responsible for the progressive hearing loss observed in subjects with WS. Electromotile amplitude could be not affected, reduced (as in the case of LIMK inhibition or deficiency), or even increased, contributing to exacerbate hyperacusis.
Thus, the reported role of LIMK in the regulation of OHC motile responses provides a strong support to the hypothesis that hyperacusis in subjects with WS would be linked to deficiency in LIMK1. In addition, the potential involvement of LIMK1 and OHC motility in the phenomenon of hyperacusis provides a consistent and strong frame of reference for understanding the cellular and molecular mechanisms involved in the regulation of the cochlear amplifier.
Figures and Tables
Figure 1 A novel model of gain-control for cochlear amplification based on the regulation of slow motility of cochlear outer hair cells. (A) The organ of Corti and related structures at a given steady-state condition. D, distance between the lower surface of the tectorial membrane and the tip of IHCs' stereocilia; L, total OHC length. (B) Proposed effect of ACh. OHCs would elongate (LACh>L) and D would increase (DACh>D). Auditory gain would decrease. (C) Proposed effect of LIMK1 deficiency in WS. OHCs would shorten (LWS<L) and D would decrease (DWS<D). Auditory gain would increase (hyperacusis). Changes in OHC length are exaggerated for clarity. See text for details.
Acknowledgements
This work was supported by National Institutes of Health grant No. DC010146 and House Ear Institute. Its content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the House Ear Institute.
Addendum to:
References
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet 1993; 5:11 - 16
- Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays 2004; 26:141 - 150
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet 2001; 29:321 - 325
- Morris CA. Introduction: Williams syndrome. Am J Med Genet C Semin Med Genet 2010; 154:203 - 208
- Marler JA, Sitcovsky JL, Mervis CB, Kistler DJ, Wightman FL. Auditory function and hearing loss in children and adults with Williams syndrome: cochlear impairment in individuals with otherwise normal hearing. Am J Med Genet C Semin Med Genet 2010; 154:249 - 265
- Zarchi O, Attias J, Gothelf D. Auditory and visual processing in williams syndrome. Isr J Psychiatry Relat Sci 47:125 - 131
- Gothelf D, Farber N, Raveh E, Apter A, Attias J. Hyperacusis in Williams syndrome: characteristics and associated neuroaudiologic abnormalities. Neurology 2006; 66:390 - 395
- Johnson LB, Comeau M, Clarke KD. Hyperacusis in Williams syndrome. J Otolaryngol 2001; 30:90 - 92
- Baguley DM. Hyperacusis. J R Soc Med 2003; 96:582 - 585
- Paglialonga A, Barozzi S, Brambilla D, Soi D, Cesarani A, Gagliardi C, et al. Cochlear active mechanisms in young normal-hearing subjects affected by Williams syndrome: Time-frequency analysis of otoacoustic emissions. Hear Res 2011; 272:157 - 167
- Tognola G, Grandori F, Avan P, Ravazzani P, Bonfils P. Frequency-specific information from click evoked otoacoustic emissions in noise-induced hearing loss. Audiology 1999; 38:243 - 250
- Jedrzejczak WW, Blinowska KJ, Konopka W. Time-frequency analysis of transiently evoked otoacoustic emissions of subjects exposed to noise. Hear Res 2005; 205:249 - 255
- Guinan JJ. Dallos P, Popper AN, Fay RR. Physiology of Cochlear Efferents. The Cochlea 1996; New York Springer-Verlag 435 - 502
- Taranda J, Maison SF, Ballestero JA, Katz E, Savino J, Vetter DE, et al. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol 2009; 7:18
- Zarchi O, Attias J, Raveh E, Basel-Vanagaite L, Saporta L, Gothelf D. A Comparative Study of Hearing Loss in Two Microdeletion Syndromes: Velocardiofacial (22q11.2 Deletion) and Williams (7q11.23 Deletion) Syndromes. J Pediatr 2011; 158:301 - 306
- Meyer J, Furness DN, Zenner HP, Hackney CM, Gummer AW. Evidence for opening of hair-cell transducer channels after tip-link loss. J Neurosci 1998; 18:6748 - 6756
- Osborne MP, Comis SD. Action of elastase, collagenase and other enzymes upon linkages between stereocilia in the guinea-pig cochlea. Acta Otolaryngol 1990; 110:37 - 45
- Preyer S, Hemmert W, Zenner HP, Gummer AW. Abolition of the receptor potential response of isolated mammalian outer hair cells by hair-bundle treatment with elastase: a test of the tip-link hypothesis. Hear Res 1995; 89:187 - 193
- Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, et al. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 2004; 428:950 - 955
- Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, et al. The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci 2006; 26:7022 - 7034
- Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 2007; 449:87 - 91
- Sakaguchi H, Tokita J, Muller U, Kachar B. Tip links in hair cells: molecular composition and role in hearing loss. Curr Opin Otolaryngol Head Neck Surg 2009; 17:388 - 393
- Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med 2007; 85:555 - 568
- Meng Y, Zhang Y, Tregoubov V, Falls DL, Jia Z. Regulation of spine morphology and synaptic function by LIMK and the actin cytoskeleton. Rev Neurosci 2003; 14:233 - 240
- Matsumoto N, Kitani R, Maricle A, Mueller M, Kalinec F. Pivotal role of actin depolymerization in the regulation of cochlear outer hair cell motility. Biophys J 2010; 99:2067 - 2076
- Lukashkin AN, Richardson GP, Russell IJ. Multiple roles for the tectorial membrane in the active cochlea. Hear Res 2009; 266:26 - 35
- Nowotny M, Gummer AW. Nanomechanics of the subtectorial space caused by electromechanics of cochlear outer hair cells. Proc Natl Acad Sci USA 2006; 103:2120 - 2125
- Chiaradia C, Nowotny M, Gummer AW. Cooper NP, Kemp DT. Deflection of IHC stereocilia in response to OHC somatic motility. Concepts and Challenges in the Biophysics of Hearing 2009; New Jersey, London, Singapur World Scientific Publishing Company 283 - 287