Abstract
High-throughput studies have enabled the large-scale mapping of synthetic lethal genetic interaction networks in the budding yeast Saccharomyces cerevisiae (S. cerevisiae). Recently, complementary high-throughput methods have been developed to map genetic interactions in the fission yeast Schizosaccharomyces pombe (S. pombe), enabling comparative analyses of genetic interaction networks between S. pombe and S. cerevisiae, two species separated by hundreds of millions of years of evolution. The resultant data has providing our first view of a possible core genetic interaction network shared between two distantly related eukaryotes, and identified numerous species-specific interactions that may contribute to the unique biology of these two different organisms. These and other results suggest that comparative interactomic studies will provide novel insights into the structure of genetic interaction networks.
High-throughput genetic interaction mapping projects seek to identify all the genetic interactions between a given query gene and a set (or ‘array’) of hundreds or thousands of target genes. Various combinations of query and array gene function have been examined, including null (both homozygous and heterozygous) and hypomorphic gene mutations, as well as overexpression of wild-type genes.Citation1–Citation6 These data can be used to identify genes that function in common, opposing or compensatory pathways, predict the function of uncharacterized gene products on the basis of their spectrum of interactions and infer the composition of multi-protein complexes.Citation3,Citation7–Citation9 There is substantial interest in improving the performance of existing systems, developing new methods and systems to examine genetic interactions in different organisms and combining the resultant knowledge to expand our understanding of eukaryotic cell biology.
Yeast Leads the Way
High-throughput mapping of genetic interactions was pioneered in the budding yeast S. cerevisiae, where the Synthetic Genetic Array (ScSGA) analysis technique was developed to investigate genetic interactions between a given query gene mutation and each of the ∼4,700 nonessential genes.Citation3,Citation10 In this technique, two parental strains harboring single gene deletions are manipulated to create recombinant double mutant progeny. The growth of the double mutant colony is compared to the growth of the individual single mutant colonies to identify those combinations of mutations that display a greater than expected (synthetic) fitness defect manifested by completely defective (lethal) or slow growth (sick) phenotypes. This method is readily automated and the requisite colony manipulations can be performed on a solid agar surface at high densities (1,536 individual colonies per plate is standard). ScSGA synthetic lethal analysis has been used to study the pathways regulating secretion, sister chromatid cohesion, DNA synthesis and repair and tRNA export, as well as to elucidate the global architecture of genetic interaction networks.Citation7,Citation10–Citation14 In addition to ScSGA, several related methods have been developed that also enable pairwise genetic interactions to be mapped systematically in S. cerevisiae.Citation15–Citation17 Moreover, conceptually similar large-scale methods have been used to examine genetic interaction networks in a multi-cellular organism, the nematode worm C. elegans.Citation4,Citation5 In these approaches, RNA interference (RNAi) is used to inactivate the function of one or both genes under study. The future application of RNAi-based methodologies to study genetic interactions in mammalian cell culture models is eagerly anticipated.
Despite advances in the mapping of genetic interactions in metazoan organisms, single-celled yeasts remain the premier model system for high-throughput genetic network mapping for three reasons. First, the existence of genome-wide deletion libraries that specifically eliminate the function of a target gene ensures that all observed phenotypes in the corresponding mutant are ‘on target’ and thus highly reproducible. Second, simple, automated methods to manipulate yeast strains enable massive parallelization and truly genome-wide datasets to be collected efficiently. Third, the emergence of quantitative methods to evaluate mutant phenotypes permit the detection and classification of double mutant phenotypes as additive, synthetic or epistatic.Citation18–Citation21 Together, these advantages enable the high confidence and high throughput detection of subtle genetic interactions that would be misclassified or elude detection altogether ‘by eye’ alone.
Building upon the conceptual and technical advances made in S. cerevisiae, new SGA-like techniques have emerged that enable high-throughput analysis of genetic interactions in other single-celled species including the bacterium E. coli (eSGA and GIANT-Coli)Citation22,Citation23 and the fission yeast Schizosaccharomyces pombe (PEM and SpSGA).Citation18,Citation24,Citation25 These methods open the way to large-scale ‘comparative interactomic’Citation26,Citation27 studies of genetic networks between different species.
High-Throughput Comparative Analysis of Genetic Interactions
Some genes are common to most if not all eukaryotes,Citation28 while others are specific to different branches of the evolutionary tree. Similarly, one would predict that certain genetic interactions between genes will be broadly conserved between species while others will be specific to, and help define, a given evolutionary sub-group. The combinatorial possibilities inherent in genetic (and protein-protein) interaction networks suggests that the way genes and gene products are wired together is likely as important in defining cell function as the function of individual genes themselves. Thus, an important goal is to define conserved and species-specific (as well as family, genus, phylum-specific, etc.,) genetic interaction networks.
The first experimental comparisons of genetic interaction networks were made between S. cerevisiae and C. elegans. Intersection of the high-throughput genetic interaction networks for these two organisms found less than 5% conservation.Citation4,Citation5,Citation29 On the other hand, a recent smaller-scale study determined that a significant fraction (9/21 or 43%) of tested genetic interactions between genes involved in mitotic spindle assembly are conserved between S. cerevisiae and C. elegans.Citation30 One possibility is that the conservation of genetic interactions between species is limited to certain processes, such as spindle formation, even when the genes for other processes are conserved. It is also possible that previous comparisons may have failed to turn up more significant degree of conservations between species for technical reasons (discussed in ref. Citation18).
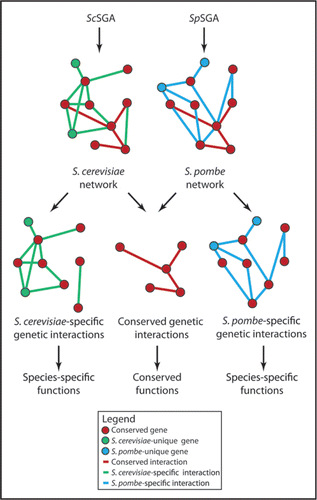
We were recently able to begin addressing these questions through a comparative study of genetic interactions in two different yeast species, S. cerevisiae and S. pombe. These two species are separated by ∼1,000 million years of evolutionCitation31 and both offer all the technical advantages of single-celled organisms with respect to the quantitative mapping of genetic interaction networks (see above). First, we performed a large-scale curation of the S. pombe literature to catalogue all known genetic interactions for this organism. By integrating this information with an existing, independent literature curation effortCitation32 we were able to determine that 23% of literaturecurated synthetic sick/synthetic lethal (SS/SL) genetic interactions were conserved between S. pombe and S. cerevisiae.Citation18 We then developed a new method, which we call S. pombe Synthetic Genetic Analysis (SpSGA), that enables mutants harboring null alleles of two non-essential genes to be isolated rapidly and scored quantitatively in this organism.Citation18 We applied both ScSGA and SpSGA methods to examine genetic interactions between a matrix of approximately 225 × 225 orthologous genes involved in a wide variety of biological processes in these two divergent yeasts (). Similar methods were also applied to a distinct set of S. pombe and S. cerevisiae genes by another group.Citation24 Importantly, both studies report similar results with respect to the overall number of conserved synthetic sick/synthetic lethal (SS/SL) genetic interactions, on the order of ∼30%. Together, these results support the hypothesis that a significant number of genetic interactions are conserved between these distantly related species, consistent with previous observations that 65% of S. cerevisiae essential genes retain their essential function in S. pombe.Citation33 Nevertheless, it is clear from these results that the majority of SS/SL genetic interactions and a significant number of essential genes are species-specific, implying substantial rewiring of the genetic interaction network of these two species. Approximately 75% of S. pombe genes have a recognizable ortholog in S. cerevisiae (S. pombe GeneDB, www.genedb.org/genedb/pombe/). Thus, to a first approximation, these data suggest that significant diversity between single-celled organisms may be generated throughout evolution by rearranging the ‘wiring’ between genes.
The field of comparative genetic interaction analysis is in its infancy and the results obtained to date raise many questions for which there are currently no good answers. First, how and why do species-specific genetic interactions arise between genes that are broadly conserved ( and reviewed in ref. Citation18)? Are these interactions truly species-specific, or simply context-dependent? For example, it is known that certain genetic interactions may only become apparent under particular nutrient conditions,Citation34 and therefore it is possible that assaying genetic interactions under different conditions (e.g., in more naturalistic environments) will reveal differences in the rate of conservation. Crucially, how do species-specific genetic interactions relate to species-specific biology? Second, how are species-specific genes integrated into conserved genetic interaction networks formed between highly conserved genes ( and reviewed in ref. Citation18)? Third, do certain classes of genes, whether defined by functional category, sequence conservation, or some other measure, retain their interactions to a greater degree than other classes? Fourth, how does the degree of network overlap correlate with evolutionary distance? Finally, can we use our knowledge of conserved and species-specific genetic interactions either alone, or combined with other types of comparative data (co-expression, protein-protein interactions, etc.,),Citation35,Citation36 to predict whether or not two genes are likely to interact with one another in any given species? To answer these questions and others we will require more comprehensive coverage of genetic interaction networks in S. cerevisiae and S. pombe, the examination of genetic interactions in other yeast species, and the further development of techniques to map genetic interactions rapidly in key metazoan model organisms such as C. elegans, Drosophila, M. musculus and human tissue culture models.
Conclusions
In the near term, genetic interactions documented in simple, experimentally tractable model organisms such as yeast will improve our understanding of basic eukaryotic cell function, reveal how conserved gene products are functionally interconnected, show how this wiring is rearranged during evolution to generate different cell types and organisms and help predict genetic interactions in more complex species. In the longer term, a better understanding of conserved and species-specific genetic interaction networks could allow us to infer genetic networks for ancestral or extinct species, improve our ability to rationally modify existing organisms for biotechnology purposes and, potentially, contribute to the design of synthetic organisms containing the appropriate genetic ‘wiring’ necessary to sustain life or execute a given task.
Figures and Tables
Figure 1 A ball and stick representation of the interactions (edges) between different genes (nodes) in two model genetic interaction networks: one for S. cerevisiae generated using the ScSGA method and one for S. pombe generated using SpSGA. Conserved interactions are presumed to contribute to processes shared between both species, while species-specific interactions contribute to species-specific functions; however, this remains to be demonstrated.
Acknowledgements
This work was supported by a postdoctoral fellowship from the Terry Fox Foundation of the National Cancer Institute of Canada to Scott J. Dixon and grants from Genome Canada (2004-OGI-3-01) and the Canadian Institutes of Health Research (GSP-41567) to Brenda Andrews and Charles Boone.
Addendum to:
References
- Haarer B, Viggiano S, Hibbs MA, Troyanskaya OG, Amberg DC. Modeling complex genetic interactions in a simple eukaryotic genome: actin displays a rich spectrum of complex haploinsufficiencies. Genes Dev 2007; 21:148 - 159
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 2006; 21:319 - 330
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001; 294:2364 - 2368
- Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet 2006; 38:896 - 903
- Byrne AB, Weirauch MT, Wong V, Koeva M, Dixon SJ, Stuart JM, et al. A global analysis of genetic interactions in Caenorhabditis elegans. J Biol 2007; 6:8
- Davierwala AP, Haynes J, Li Z, Brost RL, Robinson MD, Yu L, et al. The synthetic genetic interaction spectrum of essential genes. Nat Genet 2005; 37:1147 - 1152
- Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet 2007; 8:437 - 449
- Phillips PC. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 2008; 9:855 - 867
- Collins SR, Miller KM, Maas NL, Roguev A, Fillingham J, Chu CS, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 2007; 446:806 - 810
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, et al. Global mapping of the yeast genetic interaction network. Science 2004; 303:808 - 813
- Setty SR, Strochlic TI, Tong AH, Boone C, Burd CG. Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat Cell Biol 2004; 6:414 - 419
- Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, et al. Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell 2004; 15:1736 - 1745
- Chang M, Bellaoui M, Zhang C, Desai R, Morozov P, Delgado-Cruzata L, et al. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J 2005; 24:2024 - 2033
- Hurto RL, Tong AH, Boone C, Hopper AK. Inorganic phosphate deprivation causes tRNA nuclear accumulation via retrograde transport in Saccharomyces cerevisiae. Genetics 2007; 176:841 - 852
- Pan X, Yuan DS, Xiang D, Wang X, Sookhai-Mahadeo S, Bader JS, et al. A robust toolkit for functional profiling of the yeast genome. Mol Cell 2004; 16:487 - 496
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 2005; 123:507 - 519
- Decourty L, Saveanu C, Zemam K, Hantraye F, Frachon E, Rousselle JC, et al. Linking functionally related genes by sensitive and quantitative characterization of genetic interaction profiles. Proc Natl Acad Sci USA 2008; 105:5821 - 5826
- Dixon SJ, Fedyshyn Y, Koh JL, Prasad TS, Chahwan C, Chua G, et al. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc Natl Acad Sci USA 2008; 105:16653 - 16658
- Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol 2006; 7:63
- Mani R, St Onge RP, Hartman JLt, Giaever G, Roth FP. Defining genetic interaction. Proc Natl Acad Sci USA 2008; 105:3461 - 3466
- St Onge RP, Mani R, Oh J, Proctor M, Fung E, Davis RW, et al. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat Genet 2007; 39:199 - 206
- Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods 2008; 5:781 - 787
- Butland G, Babu M, Díaz-Mejía JJ, Bohdana F, Phanse S, Gold B, et al. eSGA: E. coli synthetic genetic array analysis. Nat Methods 2008; 5:789 - 795
- Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science 2008; 322:405 - 410
- Roguev A, Wiren M, Weissman JS, Krogan NJ. High-throughput genetic interaction mapping in the fission yeast Schizosaccharomyces pombe. Nat Methods 2007; 4:861 - 866
- Sharan R, Ideker T. Modeling cellular machinery through biological network comparison. Nat Biotechnol 2006; 24:427 - 433
- Kiemer L, Cesareni G. Comparative interactomics: comparing apples and pears?. Trends Biotechnol 2007; 25:448 - 454
- Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002; 415:871 - 880
- Tischler J, Lehner B, Fraser AG. Evolutionary plasticity of genetic interaction networks. Nat Genet 2008; 40:390 - 391
- Tarailo M, Tarailo S, Rose AM. Synthetic lethal interactions identify phenotypic “interologs” of the spindle assembly checkpoint components. Genetics 2007; 177:2525 - 2530
- Hedges SB. The origin and evolution of model organisms. Nat Rev Genet 2002; 3:838 - 849
- Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res 2008; 36:637 - 640
- Decottignies A, Sanchez-Perez I, Nurse P. Schizosaccharomyces pombe essential genes: a pilot study. Genome Res 2003; 13:399 - 406
- Harrison R, Papp B, Pal C, Oliver SG, Delneri D. Plasticity of genetic interactions in metabolic networks of yeast. Proc Natl Acad Sci USA 2007; 104:2307 - 2312
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003; 302:249 - 255
- Gandhi TK, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, et al. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 2006; 38:285 - 293