Abstract
Alzheimer’s disease (AD) is a human neurodegenerative disease characterized by co-existence of extracellular senile plaques (SP) and neurofibrillary tangles (NFT) associated with an extensive neuronal loss, primarily in the cerebral cortex and hippocampus. Several studies suggest that caspase(s)-mediated neuronal death occurs in cellular and animal AD models as well as in human brains of affected patients, although an etiologic role of apoptosis in such neurodegenerative disorder is still debated. This review summarizes the experimental evidences corroborating the possible involvement of apoptosis in AD pathogenesis and discusses the usefulness of ad hoc devised in vitro approaches to study how caspase(s), amyloidogenic processing and tau metabolism might reciprocally interact leading to neuronal death.
Introduction
Alzheimer disease (AD) is the most common human late-onset and sporadic neurodegenerative disorder characterized by global cognitive decline including a progressive loss of memory, orientation and reasoning. The neuropathological hallmarks of AD include synaptic loss and/or dysfunction, diminished neuronal metabolism, senile plaques (SPs), neurofibrillary tangles (NFTs) and loss of multiple neurotransmitter systems.Citation1
SPs typically consist of aggregated amyloid beta (Aβ), abnormal neurites and glial cells. The accumulation of Aβ due to a dysregulated proteolytic processing of its precursor molecule, the Amyloid Precursor Protein (APP), exerts a crucial role in neuronal loss or dysfunction through a cascade of events which include oxidative stress, membrane damage, altered mitochondrial metabolism, abortive cell cycle events, Ca++ imbalance, protein misfolding, DNA damage/repair and inflammatory processes.Citation2
NFTs are intracellular accumulations of cytoskeletal elements, largely made of Paired Helical Filaments (PHF), whose main constituent is abnormally phosphorylated tau. Tangles could potentially damage neurons by disrupting transport of various cellular components, including that of Nerve Growth Factor (NGF)-receptor complex, thus leading to degeneration of the tangle-bearing neurons.Citation3
Thanks to the experimental work carried out in hundreds of laboratories, it has been unequivocally demonstrated that both APP and tau proteins play a crucial role in the onset of AD. Moreover, several strong genetic evidences corroborate the “amyloid cascade hypothesis” according to which Aβ production is the trigger factor affecting downstream tau metabolism.Citation4 Mutations in several known genes linked to AD familial forms (APP, presenilin-1 or presenilin-2 gene) and genetic or environmental risk factors (Apolipoprotein E 4 variant and metals or pesticides exposure) alter Aβ cellular processing or its properties, leading to an increase of the Aβ42/40 ratio or its propensity to aggregate.Citation1 Moreover, Aβ causes caspases-mediated tau cleavage and hyperphosphorylation by activating specific kinases, thus promoting its aggregation, mis-localization and accumulation with consequent NFTs formation.Citation5
Although it is still unclear why specific vulnerable neuronal population, with special emphasis to forebrain cholinergic neurons which provide the majority of cholinergic innervations to cerebral cortex and hippocampus, die in the brain of AD patients, a growing number of studies actually indicate apoptosis as possible initial trigger of the pathology.Citation6,Citation7
In this review we will summarize the current findings regarding this hypothesis and we will discuss the convenience of ad hoc devised in vitro models to dissect the single molecular steps linking apoptosis with Aβ production and tau altered processing. A special emphasis will be devoted to analyze the possible crucial role of NGF and other neurotrophins, since the evidences demonstrating its involvement in the onset of AD are becoming conspicuous.Citation8
Alzheimer Disease and Apoptotic Events
Several studies presently indicate that apoptosis might occur in, and contribute to, AD onset and progression.Citation7 Stimuli for apoptosis in AD include increased oxidative stress, dysregulation of ion homeostasis, growth factor deprivation, accumulation of Aβ, metabolic impairment, reduced clearance of toxin, mitochondrial dysfunction, DNA damage, protein aggregation.Citation9,Citation7 Nevertheless, while the role of apoptosis in in vitro models and transgenic animal models of neuro-degeneration has been largely documented, its occurrence and role in human postmortem AD brain is controversial. Despite a growing number of studies underlying caspases and apoptosis involvement in AD, no direct role of apoptotic death in AD etiology has still been proven although the presence of apoptotic bodies, DNA fragmentation, granulated and marginated chromatin and shrunken and irregular cell shapes have been largely reported in tissue sections of brains from affected patients.Citation10,Citation11 Moreover, an imbalanced level of some molecular apoptotic markers such as pro-apoptotic (Bax, Bak and Bad) and anti-apoptotic (Bcl-2 and Bcl-xL) proteins—members of Bcl-2 protein familyCitation12,Citation13—and the initiator caspases 8 and 9 and the effector caspases 3 and 6 have been also reported in post-mortem brains of AD patients.Citation11,Citation14–Citation20 Moreover, expression profiling analysis of thousands of genes in brain tissue samples from AD and age-matched control patients has revealed a marked decrease in expression of some anti-apoptotic gene such as NCKAP1.Citation21 In addition, immunohistochemical and biochemical studies report the presence of active caspase(s) and caspase-cleaved substrates in neurons, around senile plaques and neurofibrillary tanglesCitation10,Citation11,Citation22,Citation23 and also in postsynaptic densities.Citation24 Both caspase-cleaved APP and activated caspase 3 have been shown to be present and associated to granulovacuolar degeneration, a diagnostic AD neuropathological sign in brains of affected patients.Citation25 Finally, a marked co-localization of pathological hyperphosphorylated tau, cleaved caspase-3 and caspase-6 have been recently reported in TUNEL-positive neurons in the brainstem of AD patients.Citation26
Caspases and APP.
Caspases have a direct role in amyloid precursor protein (APP) processing and in the biogenesis of Aβ peptide species.Citation27 Particularly, the C31 C-terminal peptide obtained by caspase-3 mediated APP cleavage seems to mediate apoptosis by transcriptional regulation of some genes.Citation28 Caspase-3 mediated APP cleavage also stabilizes BACE—the β-secretase enzyme initiating the APP cleavage to produce Aβ peptide—which accumulates in endosomes, where increases Aβ production.Citation29
Exposure of cultured cortical neurons to Aβ or infection of rat hippocampal neurons with APP-expressing adenovirus which causes an Aβ accumulation, induces activation of capsase-3 and apoptosis,Citation30–Citation33 suggesting that caspase(s) not only participate in the generation of Aβ but they may also directly mediate its toxic effect on neuronal survival.Citation34
As will be discussed below, APP-derived toxic peptides may not only originate by apoptosis activation but may also be responsible of it in viable neurons. Thus, APP-derived Aβ peptides can activate caspases through the extrinsic pathway, implicating binding of extracellular Aβ to cell sites, while other studies suggest that the intrinsic pathway may be more relevant.Citation35 Intracellular accumulation of Aβ in endosplasmic reticulum or endosomes may activate apoptotic mechanism(s) through the unfolded protein response (URP) or endoplasmatic reticulum stress.Citation36 Alternatively, intracellular Aβ may bind to alcohol dehydrogenase within mitochondria and activates apoptosis causing mitochondrial stress.Citation37 Interaction of Aβ with mitochondrial Cyclophilin D causes synaptic damage observed in AD and absence of Cyclophilin D protects neurons from Aβ- and oxidative stress-induced apoptotic cell death.Citation38 Aβ 1–42 also impairs proteasome activity and Aβ immunotherapy rescues the proteasome dysfunction reported in 3X transgenic AD animal models thus confirming that its intracellular accumulation alters the ubiquitin-proteasomal system in vivo.Citation39 Aβ upregulates the intracellular levels of E2-25K/Hip-2, an E2 ubiquitin-conjugating enzyme, which stabilizes endoplasmic reticulum (ER)-resident caspase-12 protein by inhibiting proteasome activity.Citation40
Pharmacological or molecular inhibition of particular members of the caspases family, such as caspase 2, 3, 8 and 12 has been reported to offer partial or complete protection against Aβ-induced apoptotic cell death in vitro.Citation41–Citation45
As far as the effect of caspase(s) inhibition on APP metabolism in cellular and animal models, it has been reported that specific downregulation of caspase-6 in human primary neuronal cultures prevents serum-deprivation mediated Aβ increase, as well as in vitro cell death.Citation46 In a similar fashion, IETD, a caspase III inhibitor including caspase-6, -8 and -9 prevents APP cleavage in staurosporine-induced cell death in COS transfected cells.Citation47 In agreement with previous in vitro experimental data, caspase inhibition in vivo by bafilomicin, a pan-caspase(s) inhibitor, abolishes brain trauma-induced increase in Aβ and reduces neuronal degeneration in hippocampus of injected mice.Citation48 Finally, it is noteworthy that in vivo inhibition of cathepsin B improves memory and synaptic transmission in transgenic mice overexpressing APP, interfering with amyloidogenic APP processing.Citation49 On the contrary, calpain inhibition is also protective in vivo against cognitive loss in another AD animal model-APP/PS1 mice- by upregulating the phosphorylation levels of the transcription factor CREB (cAMP Responsive Element Binding Protein) without any significant change in Aβ peptides levels.Citation50
Caspases and tau.
Studies from cellular and animal models indicate that caspases have also been implicated in mechanisms of tau-mediated neurodegeneration in AD.Citation51,Citation52 According to this hypothesis, Aβ peptide promotes neuronal pathological tau filament assembly by triggering caspases activation leading to tau cleavage.Citation53 This event, in turn, generates a proteolytic products that assemble more rapidly and extensively into tau pathological filaments.Citation54,Citation55 Aberrant activation of caspase(s), following apoptotic stimuli or neurodegenerative insults, may produce one or more toxic NH2-tau fragments, that further contribute to propagate and increase cellular dysfunctions in AD.Citation56,Citation57 Colocalization of hyperphosphorylated tau and active caspase-3 and 6 has been recently detected in brainstem of young and old AD patients.Citation26 The finding that the rTg4510 tau transgenic mouse shows caspase-3 activation provides additional supporting evidence linking caspase-3 and tau-mediated neurodegeneration.Citation58 Caspase-9 activation and caspase-cleaved tau forms have been documented in AD hippocampal brain sections.Citation18 Finally overexpression of Bcl-2 in a triple transgenic Alzheimer mouse model harboring PS1(M146V), APP(Swe) and tau(P301L) transgenes limits caspase-9 activation, attenuates the processing of APP and tau thus reducing the number of NFTs and extracellular deposits of Aβ associated with the progression of this disease.Citation59
It remains to be determined if frank apoptosis is a necessary and early event in the neurodegeneration. According to this view, a positive feedback loop in neurodegeneration would be activated whereby caspase(s) generate Aβ, which in turn exerts a noxious action on tau proteins and further activates caspase(s) in neighboring neurons eventually dying by apoptosis. In this context, other modes of cell death could contribute to neuronal loss in ADCitation60 and other proteases, such as calpain and cathepsin, can be also directly or indirectly activated by caspases during apoptosis.Citation61 Finally, an intricate cross-talk between these proteases systems has been reported during apoptosis of neuronal cells.Citation62 Thus, although other caspase-independent pathways may contribute to the AD progression, the in vivo treatment with specific caspase(s) inhibitors, which are able to penetrate the blood-brain barrier, may still offer an useful and alternative therapeutic strategy to delay selective neuronal loss associated to such neurodegenerative disease.
The Cerebellar Granule Cells (CGC) Model
A decade ago, our research group hypothesized a possible tight link between improper activation of apoptosis and events related to AD. Cerebellar Granule Neurons (CGNs) from 8 days old rat require depolarizing potassium concentration (25 mM K+) for an optimal survival, when explanted in vitro. Upon reduction of extracellular potassium concentration to a more physiological concentration of 5 mM, these neurons progressively undergo apoptosisCitation63 which is largely blocked by neuroprotective agents able to increase calcium influx.Citation64 It has been hypothesized that in vitro depolarizing conditions are necessary to maintain intracellular high levels of free calcium, thus mimicking the in vivo situation of continuous electrical stimulation related to the developmental establishment of excitatory synapses originating from mossy fibers.Citation9,Citation65 The apoptotic process, as well as nuclear and mitochondrial damage, are reversible up to 4–8 hours of induction suggesting that no rescue is possible even if CGNs are returned to high K+ medium.Citation66,Citation67 Activation of caspase-3 has been reported after serum/K+ starvationCitation68 and cell death is attenuated by the selective caspase-3 inhibitor z-DEVD-fmk;Citation69,Citation70 although the main effect of such caspase is on DNA fragmentation and chromatin condensation rather than preventing apoptosis.Citation71 Such conflicting data may reflect the finding that neuronal apoptosis triggered by potassium reduction involves a more intricate caspase(s) activation cascadeCitation72 and a cross-talk between caspase(s) and other protease(s) further complicates the death signaling.Citation73–Citation76 Neurotrophin and physiological neuropeptides, such as IGF, bFGF, BDNF, PACAP, SP and cAMPCitation63,Citation64,Citation76–Citation79 also exert their protective action in this neuronal paradigm through different mechanism including the activation of PI3-kinase/Akt pathway;Citation76,Citation78,Citation80,Citation81 the stimulation of PKA and/or MAP kinases signaling.Citation82,Citation83
We have been reported that the pro-apoptotic shifting to a low potassium medium activates an amyloidogenic process, which rapidly and irreversibly leads to an unbalance between the “physiological” α-secretase-mediated pathway and the β-α-secretase mediated increased production of Aβ.Citation84 Moreover, the monomeric and oligomeric forms of 4-kDa Aβ are significantly higher in depolarization-stimulated secretion compared with controls. Such increments are paralleled by a corresponding increase of the β-APPs/α-APPs ratio in apoptotic secretion, without any significant change of intracellular full-length APP levels. An interesting aspect of such a process is that the released pool of Aβ may activate a toxic loop that further accelerates and propagates the process of neurodegeneration, affecting neighboring healthy neurons. Indeed, co-incubation of apoptotic cultures with antibodies directed against Aβ significantly slows down the extension of cell death and quantitatively increases the neuronal survival rate by approximately 50%,Citation85,Citation86 thus suggesting that Aβ peptides may act as soluble and diffusible apoptotic death mediators.
Contextually to the significant increase of amyloidogenic metabolism of APP, also tau undergoes post-translational modifications. After 6 h of potassium deprivation, a change in tau phosphorylation state and caspase(s) and calpain-mediated cleavage occurs in concomitance with a progressive disassembly of cytoskeleton, eventually leading to the generation of a 17 kDa fragment which accumulates in the perikarya of dying cells.Citation73 Furthermore, following the apoptotic trigger, a reactive oxygen species (ROS) production and progressively reduced mitochondrial function also contribute to neuronal damage.Citation87,Citation88 Superoxide dismutase, N-acetyl-L-cysteine and other free radical scavengers partially protect CGNs form death, improving mitochondrial energy metabolism.Citation89,Citation90
The bulk of studies on CGNs, apoptosis and events related to AD prospected a first, consistent positive answer to their possible link. Nevertheless, the observation that these neurons are not the most vulnerable population affected in AD and that few clinical signs of cerebellar anatomopathological dysfunction have been reported in AD patients leaves room for some criticisms about its fully usefulness as in vitro model for this human neurodegenerative disease.
NGF-Deprived PC12 and Hippocampal/Cortical Neuronal Models
NGF (Nerve Growth Factor) is the first neurotrophin to be discovered and is not only endowed with the property of inducing growth of nerve fibers in target neurons, but also of supporting their life via its antiapoptotic actionCitation91 Numerous in vitro and in vivo data suggest a tight causal relationship between an imbalance in NGF receptor signaling, the activation of amyloidogenic pathway and altered tau metabolism in onset and progression of AD-like neurodegeneration.
TrkA, the high affinity NGF receptor, has been found decreased in the basal forebrainCitation92–Citation97 and in the cortex.Citation98–Citation100 A switch from TrkA to p75, the low affinity death receptor, it has been described during neuronal aging resulting in increased amyloidogenic processing of APP.Citation101–Citation103 p75NTR expression has been directly linked to changes occurring in AD,Citation104 including the death of basal forebrain neurons,Citation105,Citation106 hypothesized to occur through a direct binding of oligomeric Aβ1–42 peptides to p75.Citation103,Citation107,Citation108 Moreover, some evidences have previously showing a transcriptional p75-mediated regulation on the APP promoter leading to an increase of secreted amyloid precursor protein (sAPP)Citation109,Citation110 in neurons.
Several studies report a regulative role of NGF on tau phosphorylation. Stimulation of undifferentiated PC-12 with NGF causes a dephosphorylation of tau proteins,Citation111 although an increase of Gsk3β-mediated tau phosphorylation has also been observed. Interestingly, this tau phosphorylation at defined sites might be required for proper anterograde organelle/mitochondrial transport in differentiated cells.Citation112 On the other hand, NGF deprivation in differentiated PC12 induces apoptosis and hyperphosphorylation both of tau and membrane-bound high molecular weight (HMW) tau, especially in the neuritis. These changes are accompanied by an impairment of its microtubule binding ability and a marked decrease of its solubility. However, in the last stages of apoptosis, tau is dephosphorylated in dying neuronal PC12.Citation113,Citation114 In addition, in this apoptotic neuronal model, NGF deprivation also causes an early, caspase-mediated tau cleavage at NH2 domain with the appearance of the 20–22 kDa tau fragmentCitation115 which has been previously demonstrated to be markedly neurotoxic in vitro when overexpressed in primary neuronal cultures.Citation116 NGF might control the endogenous tau protein levels, regulating its metabolism via proteasomal degradation, as demonstrated by NGF-dependent ubiquitination of tau in cultured cells.Citation117 Finally, several evidences support the hypothesis that the role of tau in axonal transport might affect NGF-TrkA signaling in vivo. Indeed, experimental data from retrograde labeling of basal forebrain neurons after injection of fluorogold into multiple sites in cortex and hippocampus, report that an altered compartmentalization of phosphotau, GSK3 and TrkA immunoreactivity may be responsible for the failure of axonal trafficking and lack of trophic support in aged cholinergic cells.Citation118,Citation119
The hypothesis that a chronic NGF deprivation may be one of the factors involved in the etiology of sporadic forms of AD is validated by the findings that acute treatment with NGF or acetylcholine esterase (AChE) inhibitors, such as ganstigmine and donepezil, rescues the cholinergic and behavioral deficit in AD11 mice. These mice are an in vivo AD transgenic model, in which the phenotypic knockout of NGF is achieved by the expression of recombinant neutralizing antibodies.Citation120,Citation121 Finally, clinical encouraging data from ongoing gene therapy trial using NGF-grafted autologous fibroblasts injected into the basal nucleus of Meynert (nbM),Citation122 further validate the rational for the therapeutic administration of human recombinant NGF in AD patients.Citation123
In view of these findings, we carried out a set of experiments in NGF-deprived differentiated PC12 cellsCitation124,Citation115 and described the crucial steps linking NGF withdrawal, activation of amyloidogenesis, tau truncation and caspase(s)-mediated execution of neuronal death. These studies have been replicated in primary hippocampal and cortical neurons showing that, upon NGF removal, the amyloidogenic pathway is activated with consequent intra and extracellular accumulation of Aβ peptides and apoptotic death. The overproduced Aβ is partly released in the culture medium, where it aggregates to form structures largely reminiscent of those forming senile plaques, and in part aggregates within neurons. All these events are prevented by β and α secretase inhibitors, by antibodies directed against Aβ peptides, or by partial silencing of APP mRNA, whereas they are mimicked by Aβ 1–42 peptide exposure. Conversely, neurons deprived of serum largely die but, although the amyloidogenic pathway is activated, the exposure to anti Aβ antibodies does not protect from apoptotic death, further suggesting that the activation of amyloidogenesis following NGF withdrawal is not a simple consequence of an apoptotic trigger but it is strictly related to lack of NGF supply.Citation125
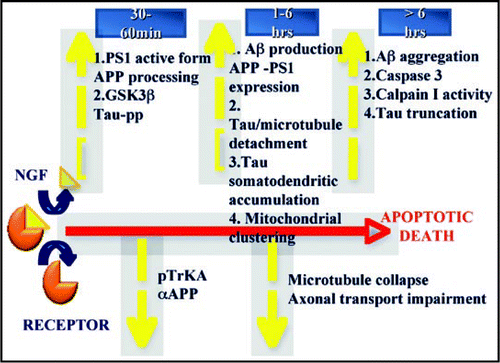
In the same experimental model we have also demonstrated an early involvement of tau protein which, under NGF deprivation, undergoes GSK3β mediated hyperphosphorylation at pathognomonic amino acids such as Ser 262 and Thr 231, and is subsequently degraded generating a toxic NH-2-derived 220 amino acid peptide.Citation116 Such tau hyperphosphorylation, as well as apoptotic death, is blocked by Aβ antibodies or by specific β and/or α-secretases inhibitors and is mimicked by Aβ 1–42 peptide, suggesting that Aβ species are the initial trigger. Tau subsequently detaches from microtubules, thus shifting the equilibrium toward its disassembled state and indirectly affecting the whole axonal transport, eventually leading to apoptotic death (Amadoro et al. submitted). Once tau is displaced from microtubules, it would be further phosphorylated at other fibrillogenic site and/or cleaved by proteases (i.e., caspase(s) and calpain), causing disruption of microtubule transport along axons and consequent synaptic dysfunction. All these events are summarized in the .
To our knowledge, the NGF-deprived hippocampal culture is presently the only in vitro model whereby both APP, tau altered processing and apoptosis, have been investigated together under strictly controlled conditions. Regarding the direct role of caspases in neuronal in vitro models whose viability is strictly dependent on NGF, an involvement of such proteases in apoptotic cell death caused by NGF deprivationCitation126,Citation127 and in the p75-mediated cell death caused by exogenous addition of Aβ to neuroblastoma cellsCitation128 has been largely documented. The studies performed in NGF-deprived PC12 cells show that, among all caspase(s) inhibitors tested, only blockage of executor caspases 2, 12, 6 and 8 exerts an almost total protection from death and from Aβ production, whereas inhibition of effector caspase 3 does not exert a similar action.Citation124 In a similar fashion and in agreement with others,Citation129,Citation130 treatment with Z-DEVD-fmk, a specific inhibitor of caspase-3 only partially rescued hippocampal neurons from death, probably because this protease is not activated at early times upon NGF withdrawal in this neuronal paradigm (Amadoro et al. submitted). On the contrary, pharmacological inhibition of caspase 3 markedly inhibited caspases-mediated tau cleavage, without any significant effects on GSK3β-mediated tau hyperphosphorylation (Amadoro et al. submitted). Moreover, the finding that the general cell-permeable caspase inhibitor z-VAD does not significantly affect ThT-positive Aβ structures production in NGF-deprived PC12, whereas partially rescues cells from apoptotic death,Citation124 delineates a complex chain of events between NGF withdrawal, Aβ production, apoptosis and tau modifications. As mentioned above, the causal and temporal relationship between caspases-mediated cell death and APP processing appears cell-specific and signaling-dependent and probably initiates a toxic cycle of cellular Aβ production/neuronal loss, which is difficult to elucidate in its actual sequence. Thus, although elevated Aβ may lead to apoptotic cell death after injury or disease and caspase(s) inhibition may protect against this event, a causal relationship could not be proven as blockade of caspase(s) might also prevent tau modifications and cell death unrelated to Aβ toxicity. Further investigations aimed at selectively reducing Aβ levels, without targeting caspase(s) activity (i.e., by directly altering α, β and/or α secretase activity), will provide additional insights into this cascade to definitively establish if apoptosis is the primary cause of Aβ production/tau modification or is it a sort of downstream consequence, eventually ending in cell death.
Figures and Tables
Figure 1 Schematic representation of the apoptotic mechanisms by which the interruption of NGF signaling affects APP processing and Tau metabolism in hippocampal neurons. For more details see text.
Acknowledgements
Financial support was received from the Regione Lazio, FIRB 2003, Prin 2006 and INRCA 2008 (Istituto Nazionale Riposo e Cura per Anziani) to P. Callisano.
References
- Selkoe DJ. Alzheimer's disease: genes, proteins and therapy. Physiol Rev 2001; 81:741 - 766
- Shen Y, He P, Zhong Z, McAllister C, Lindholm K. Distinct destructive signal pathways of neuronal death in Alzheimer's disease. Trends Mol Med 2006; 12:574 - 579
- Salehi A, Delcroix JD, Mobley WC. Traffic at the intersection of neurotrophic factor signaling and neurodegeneration. Trends Neurosci 2003; 26:73 - 80
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297:353 - 356
- Blurton-Jones M, Laferla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res 2006; 3:437 - 448
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 2000; 1:120 - 129
- Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci 2006; 7:278 - 294
- Schulte-Herbrüggen O, Braun A, Rochlitzer S, Jockers-Scherübl MC, Hellweg R. Neurotrophic factors—a tool for therapeutic strategies in neurological, neuropsychiatric and neuroimmunological diseases?. Curr Med Chem 2007; 14:2318 - 2329
- Canu N, Calissano P. In vitro cultured neurons for molecular studies correlating apoptosis with events related to Alzheimer disease. Cerebellum 2003; 2:270 - 278
- Shimohama S. Apoptosis in Alzheimer's disease—an update. Apoptosis 2000; 5:9 - 16
- Behl C. Apoptosis and Alzheimer's disease. J Neural Transm 2000; 107:1325 - 1344
- Kitamura Y, Shimohama S, Kamoshima W, Ota T, Matsuoka Y, Nomura Y, et al. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer's disease. Brain Res 1998; 780:260 - 269
- Su JH, Deng G, Cotman CW. Bax protein expression is increased in Alzheimer's brain: correlations with DNA damage, Bcl-2 expression and brain pathology. J Neuropathol Exp Neurol 1997; 56:86 - 93
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brück W, Jellinger K, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol 1999; 155:1459 - 1466
- Selznick LA, Holtzman DM, Han BH, Gökden M, Srinivasan AN, Johnson EM Jr, et al. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol 1999; 58:1020 - 1026
- Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques and neurofibrillary tangles of Alzheimer's disease. Am J Pathol 2004; 165:523 - 531
- Rohn TT, Head E, Nesse WH, Cotman CW, Cribbs DH. Activation of caspase-8 in the Alzheimer's disease brain. Neurobiol Dis 2001; 8:1006 - 1016
- Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol Dis 2002; 11:341 - 354
- Wu CK, Thal L, Pizzo D, Hansen L, Masliah E, Geula C. Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease. Exp Neurol 2005; 195:484 - 496
- Albrecht S, Bourdeau M, Bennett D, Mufson EJ, Bhattacharjee M, LeBlanc AC. Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol 2007; 170:1200 - 1209
- Suzuki T, Nishiyama K, Yamamoto A, Inazawa J, Iwaki T, Yamada T, et al. Molecular cloning of a novel apoptosis-related gene, human Nap1 (NCKAP1), and its possible relation to Alzheimer disease. Genomics 2000; 63:246 - 254
- Cribbs DH, Poon WW, Rissman RA, Blurton-Jones M. Caspase-mediated degeneration in Alzheimer's disease. Am J Pathol 2004; 165:353 - 355
- Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann Neurol 2003; 54:393 - 398
- Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA , et al. Caspase 3 is enriched in postsynaptic densities and increased in Alzheimer's disease. Am J Pathol 2008; 173:1488 - 1495
- Su JH, Kesslak JP, Head E, Cotman CW. Caspase-cleaved amyloid precursor protein and activated caspase-3 are co-localized in the granules of granulovacuolar degeneration in Alzheimer's disease and Down's syndrome brain. Acta Neuropathol 2002; 104:1 - 6
- Wai MS, Liang Y, Shi C, Cho EY, Kung HF, Yew DT. Co-localization of hyperphosphorylated tau and caspases in the brainstem of Alzheimer's disease patients. Biogerontology 2008; In press
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999; 97:395 - 406
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, et al. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med 2000; 6:397 - 404
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 2007; 54:721 - 737
- Marín N, Romero B, Bosch-Morell F, Llansola M, Felipo V, Romá J, et al. Beta-amyloid-induced activation of caspase-3 in primary cultures of rat neurons. Mech Ageing Dev 2000; 119:63 - 67
- Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured nervous system neurons. Proc Natl Acad Sci USA 1993; 90:7951 - 7955
- Uetsuki T, Takemoto K, Nishimura I, Okamoto M, Niinobe M, Momoi T, et al. Activation of neuronal caspase-3 by intracellular accumulation of wild-type Alzheimer amyloid precursor protein. J Neurosci 1999; 19:6955 - 6964
- Nishimura I, Uetsuki T, Kuwako K, Hara T, Kawakami T, Aimoto S, et al. Cell death induced by a caspase-cleaved transmembrane fragment of the Alzheimer amyloid precursor protein. Cell Death Differ 2002; 9:199 - 208
- Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res 1999; 58:167 - 190
- Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J Mol Neurosci 2001; 17:137 - 145
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003; 39:409 - 421
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004; 304:448 - 452
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 2008; 14:1097 - 1105
- Oddo S. The ubiquitin-proteasome system in Alzheimer's disease. J Cell Mol Med 2008; 12:363 - 373
- Song S, Lee H, Kam TI, Tai ML, Lee JY, Noh JY, et al. E2-25K/Hip-2 regulates caspase-12 in ER stress-mediated Abeta neurotoxicity. J Cell Biol 2008; 82:675 - 684
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403:98 - 103
- Ivins KJ, Thornton PL, Rohn TT, Cotman CW. Neuronal apoptosis induced by beta-amyloid is mediated by caspase-8. Neurobiol Dis 1999; 6:440 - 449
- Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, et al. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem 2000; 275:11553 - 11560
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci 2000; 20:1386 - 1392
- Wei Z, Song MS, MacTavish D, Jhamandas JH, Kar S. Role of calpain and caspase in beta-amyloid-induced cell death in rat primary septal cultured neurons. Neuropharmacology 2008; 54:721 - 733
- LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis and Alzheimer's disease. J Biol Chem 1999; 274:23426 - 23436
- Weidemann A, Paliga K, Dürrwang U, Reinhard FB, Schuckert O, Evin G, et al. Proteolytic processing of the Alzheimer's disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J Biol Chem 1999; 274:5823 - 5829
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, et al. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp Neurol 2006; 197:437 - 450
- Hook VY, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J Biol Chem 2008; 283:7745 - 7753
- Trinchese F, Fa' M, Liu S, Zhang H, Hidalgo A, Schmidt SD, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest 2008; 118:2796 - 2807
- Dickson DW. Apoptotic mechanisms in Alzheimer neurofibrillary degeneration: cause or effect?. J Clin Invest 2004; 114:23 - 27
- García-Sierra F, Mondragón-Rodríguez S, Basurto-Islas G. Truncation of tau protein and its pathological significance in Alzheimer's disease. J Alzheimers Dis 2008; 14:401 - 409
- Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 2005; 64:104 - 112
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA 2003; 100:10032 - 10037
- Cho JH, Johnson GV. Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J Biol Chem 2004; 279:54716 - 54723
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, et al. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 2000; 75:624 - 633
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, et al. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 2001; 8:162 - 172
- Ramalho RM, Viana RJ, Castro RE, Steer CJ, Low WC, Rodriguez CM. Apoptosis in transgenic mice expressing the P301L mutated form of human tau. Mol Med 2008; 1485:309 - 317
- Rohn TT, Vyas V, Hernandez-estrada T, Nichol KE, Christie LA, Head E. Lack of pathology in a triple transgenic mouse model of Alzheimer's disease after overexpression of the antiapoptotic protein bcl-2. J Neurosci 2008; 28:3051 - 3059
- Zilkova M, Koson P, Zilka N. The hunt for dying neurons: insight into the neuronal loss in Alzheimer's disease. Bratisl Lek Listy 2006; 107:366 - 373
- Yuan J. Divergence from a dedicated cellular suicide mechanism: exploring the evolution of cell death. Mol Cell 2006; 23:1 - 12
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature 2006; 443:796 - 802
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 1993; 90:10989 - 10993
- Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci 1995; 15:1172 - 1179
- Borsello T, Di Luzio A, Ciotti MT, Calissano P, Galli C. Granule neuron DNA damage following deafferentation in adult rats cerebellar cortex: a lesion model. Neuroscience 2000; 95:163 - 171
- Nardi N, Avidan G, Daily D, Zilkha-Falb R, Barzilai A. Biochemical and temporal analysis of events associated with apoptosis induced by lowering the extracellular potassium concentration in mouse cerebellar granule neurons. J Neurochem 1997; 68:750 - 759
- Schulz JB, Beinroth S, Weller M, Wüllner U, Klockgether T. Endonucleolytic DNA fragmentation is not required for apoptosis of cultured rat cerebellar granule neurons. Neurosci Lett 1998; 245:9 - 12
- Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci 1997; 17:553 - 562
- Eldadah BA, Yakovlev AG, Faden AI. The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells. J Neurosci 1997; 17:6105 - 6113
- Eldadah BA, Ren RF, Faden AI. Ribozyme-mediated inhibition of caspase-3 protects cerebellar granule cells from apoptosis induced by serum-potassium deprivation. J Neurosci 2000; 20:179 - 186
- D'Mello SR, Kuan CY, Flavell RA, Rakic P. Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J Neurosci Res 2000; 59:24 - 31
- Gerhardt E, Kügler S, Leist M, Beier C, Berliocchi L, Volbracht C, et al. Cascade of caspase activation in potassium-deprived cerebellar granule neurons: targets for treatment with peptide and protein inhibitors of apoptosis. Mol Cell Neurosci 2001; 4:717 - 731
- Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM, et al. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci 1998; 18:7061 - 7074
- Canu N, Tufi R, Serafino AL, Amadoro G, Ciotti MT, Calissano P. Role of the autophagic-lysosomal system on low potassium-induced apoptosis in cultured cerebellar granule cells. J Neurochem 2005; 92:1228 - 1242
- Kaasik A, Rikk T, Piirsoo A, Zharkovsky T, Zharkovsky A. Upregulation of lysosomal cathepsin L and autophagy during neuronal death induced by reduced serum and potassium. Eur J Neurosci 2005; 22:1023 - 1031
- Amadoro G, Pieri M, Ciotti MT, Carunchio I, Canu N, Calissano P, et al. Substance P provides neuroprotection in cerebellar granule cells through Akt and MAPK/Erk activation: evidence for the involvement of the delayed rectifier potassium current. Neuropharmacology 2007; 52:1366 - 1377
- Kubo T, Nonomura T, Enokido Y, Hatanaka H. Brain-derived neurotrophic factor (BDNF) can prevent apoptosis of rat cerebellar granule neurons in culture. Brain Res Dev Brain Res 1995; 85:249 - 258
- D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci 1997; 17:1548 - 1560
- Journot L, Villalba M, Bockaert J. PACAP-38 protects cerebellar granule cells from apoptosis. Ann NY Acad Sci 1998; 865:100 - 110
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997; 275:661 - 665
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857 - 868
- Kienlen Campard P, Crochemore C, René F, Monnier D, Koch B, Loeffler JP. DNA PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. Cell Biol 1997; 16:323 - 333
- Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci 1997; 17:83 - 90
- Galli C, Piccini A, Ciotti MT, Castellani L, Calissano P, Zaccheo D, et al. Increased amyloidogenic secretion in cerebellar granule cells undergoing apoptosis. Proc Natl Acad Sci USA 1998; 95:1247 - 1252
- Piccini A, Ciotti MT, Vitolo OV, Calissano P, Tabaton M, Galli C. Endogenous APP derivatives oppositely modulate apoptosis through an autocrine loop. Neuroreport 2000; 11:1375 - 1379
- De Berardinis M, Ciotti MT, Amadoro G, Galli C, Calissano P. Transfer of the apoptotic message in sister cultures of cerebellar neurons. Neuroreport 2001; 12:2137 - 2140
- Atlante A, Gagliardi S, Marra E, Calissano P. Neuronal apoptosis in rats is accompanied by rapid impairment of cellular respiration and is prevented by scavengers of reactive oxygen species. Neurosci Lett 1998; 245:127 - 130
- Atlante A, Bobba A, Calissano P, Passarella S, Marra E. The apoptosis/necrosis transition in cerebellar granule cells depends on the mutual relationship of the antioxidant and the proteolytic systems which regulate ROS production and cytochrome c release en route to death. J Neurochem 2003; 84:960 - 971
- Atlante A, Passarella S. Detection of reactive oxygen species in primary cultures of cerebellar granule cells. Brain Res Brain Res Protoc 1999; 4:266 - 270
- Schulz JB, Weller M, Klockgether T. Potassium deprivation-induced apoptosis of cerebellar granule neurons: a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J Neurosci 1996; 16:4696 - 4706
- Levi-Montalcini R. Nerve growth Factor. Science 1975; 187:113
- Boissière F, Hunot S, Faucheux B, Hersh LB, Agid Y, Hirsch EC. Trk neurotrophin receptors in cholinergic neurons of patients with Alzheimer's disease. Dement Geriatr Cogn Disord 1997; 8:1 - 8
- Chu Y, Cochran EJ, Bennett DA, Mufson EJ, Kordower JH. Downregulation of trkA mRNA within nucleus basalis neurons in individuals with mild cognitive impairment and Alzheimer's disease. J Comp Neurol 2001; 437:296 - 307
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Downregulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem 2006; 97:475 - 487
- Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer's disease. Exp Neurol 1997; 146:91 - 103
- Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, et al. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol 2000; 427:19 - 30
- Salehi A, Verhaagen J, Dijkhuizen PA, Swaab DF. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer's disease. Neuroscience 1996; 75:373 - 387
- Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer's disease. Ann Neurol 2004; 56:520 - 531
- Hock C, Heese K, Hulette C, Rosenberg C, Otten U. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol 2000; 57:846 - 851
- Savaskan E, Müller-Spahn F, Olivieri G, Bruttel S, Otten U, Rosenberg C, et al. Alterations in trk A, trk B and trk C receptor immunoreactivities in parietal cortex and cerebellum in Alzheimer's disease. Eur Neurol 2000; 44:172 - 180
- Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J 2005; 391:59 - 67
- Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid beta-peptide generation. EMBO J 2006; 25:1997 - 2006
- Costantini C, Rossi F, Formaggio E, Bernardoni R, Cecconi D, Della-Bianca V. Characterization of the signaling pathway downstream p75 neurotrophin receptor involved in beta-amyloid peptide-dependent cell death. J Mol Neurosci 2005; 25:141 - 156
- Schliebs R. Basal forebrain cholinergic dysfunction in Alzheimer's disease—interrelationship with beta-amyloid, inflammation and neurotrophin signaling. Neurochem Res 2005; 30:895 - 908
- Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, et al. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci 1997; 17:7594 - 7605
- Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci 2006; 26:7756 - 7766
- Yaar M, Arble BL, Stewart KB, Qureshi NH, Kowall NW, Gilchrest BA. p75 (NTR) antagonistic cyclic peptide decreases the size of beta amyloid-Induced brain inflammation. Cell Mol Neurobiol 2008; 28:1027 - 1031
- Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. Beta-amyloid (1–42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci 2008; 28:3941 - 3946
- Ge YW, Lahiri DK. Regulation of promoter activity of the APP gene by cytokines and growth factors: implications in Alzheimer's disease. Ann NY Acad Sci 2002; 973:463 - 467
- Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V. p75 and TrkA receptor signaling independently regulate amyloid precursor protein mRNA expression, isoform composition and protein secretion in PC12 cells. J Neurochem 1998; 71:757 - 766
- Fisher A, Heldman E, Gurwitz D, Haring R, Karton Y, Meshulam H, et al. M1 agonists for the treatment of Alzheimer's disease. Novel properties and clinical update. Ann NY Acad Sci 1996; 777:189 - 196
- Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci 2004; 117:1653 - 1663
- Nuydens R, Dispersyn G, de Jong M, van den Kieboom G, Borgers M, Geerts H. Aberrant tau phosphorylation and neurite retraction during NGF deprivation in PC12 cells. Biochem Biophys Res Commun 1997; 240:687 - 691
- Shelton SB, Johnson GV. Tau and HMW tau phosphorylation and compartmentalization in apoptotic neuronal PC12 cells. J Neurosci Res 2001; 66:203 - 213
- Corsetti V, Amadoro G, Gentile A, Capsoni S, Ciotti MT, Cencioni MT, et al. Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer's disease models. Mol Cell Neurosci 2008; 38:381 - 392
- Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci USA 2006; 103:2892 - 2897
- Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem 2005; 94:192 - 203
- Niewiadomska G, Baksalerska-Pazera M, Riedel G. Altered cellular distribution of phospho-tau proteins coincides with impaired retrograde axonal transport in neurons of aged rats. Ann NY Acad Sci 2005; 1048:287 - 295
- Niewiadomska G, Baksalerska-Pazera M, Riedel G. Cytoskeletal transport in the aging brain: focus on the cholinergic system. Rev Neurosci 2006; 17:581 - 618
- Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J, Gonfloni S, et al. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen and skeletal muscle dystrophy. J Neurosci 2000; 20:2589 - 2601
- Capsoni S, Cattaneo A. On the molecular basis linking nerve growth factor (NGF) to Alzheimer's disease. Cell Mol Neurobiol 2006; 26:619 - 633
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 2005; 11:551 - 555
- Cattaneo A, Capsoni S, Paoletti F. Towards non invasive nerve growth factor therapies for Alzheimer's disease. J Alzheimers Dis 2008; 15:255 - 283
- Matrone C, Di Luzio A, Meli G, D'Aguanno S, Severini C, Ciotti MT, et al. Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells. J Alzheimers Dis 2008; 13:81 - 96
- Matrone C, Ciotti MT, Mercanti D, Marolda R, Calissano P. NGF and BDNF signaling control amyloidogenic route and Ab production in hippocampal neurons. Proc Natl Acad Sci USA 2008; 105:13138 - 13143
- Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, et al. Multiple pathways of neuronal death induced by DNA-damaging agents, NGF deprivation and oxidative stress. J Neurosci 1998; 18:830 - 840
- Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J Neurosci 2001; 21:5007 - 5016
- Perini G, Della-Bianca V, Politi V, Della Valle G, Dal-Pra I, et al. Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid peptides and synergistic effect of inflammatory cytokines. J Exp Med 2002; 195:907 - 918 Erratum in: J Exp Med 2002; 195:1231
- Rametti A, Esclaire F, Yardin C, Terro F. Linking alterations in tau phosphorylation and cleavage during neuronal apoptosis. J Biol Chem 2004; 279:54518 - 54528
- Bhat RV, Leonov S, Luthman J, Scott CW, Lee CM. Interactions between GSK3beta and caspase signalling pathways during NGF deprivation induced cell death. J Alzheimers Dis 2002; 4:291 - 330