Abstract
Saccharomyces cerevisiae transport protein particle (TRAPP) is a family of related multisubunit complexes required for endoplasmic reticulum-to-Golgi transport (TRAPP I), endosome-to-Golgi transport (TRAPP II) or cytosol to vacuole targeting (TRAPP III). To gain insight into the relationship between these complexes, we generated random and targeted mutations in the Trs23p core subunit. Remarkably, at physiological salt concentrations only two peaks (TRAPP I and a high molecular weight peak) are detected in wild-type cells. As the salt was raised, the high molecular weight peak resolved into TRAPP II and III peaks. Deletion of a Saccharomycotina-specific domain of Trs23p resulted in destabilization of TRAPP I but had no effect on TRAPP II or III. This mutation had no observable growth phenotype, normal levels of Ypt1p-directed guanine nucleotide exchange factor activity in vivo and did not display any in vivo nor in vitro blocks in membrane traffic. Biochemical analysis indicated that TRAPP I could be produced from the TRAPP II/III peak in vitro by increasing the salt concentration. Our data suggest that the SMS domain of Trs23p is responsible for the in vitro appearance of TRAPP I in S. cerevisiae. The implications of these findings are discussed.
Introduction
Newly synthesized proteins destined for secretion are packaged into COP II-coated transport vesicles at the endoplasmic reticulum (ER).Citation1 These vesicles are then recognized by and fuse with the earliest (cis) Golgi compartment. Similar budding, recognition and fusion events take place from nearly every endomembrane compartment within the cell, and understanding how each vesicle is specifically recognized has represented a major focus of efforts in the field of vesicle transport. Many factors participate in the overall process including SNAREs, Ypt/Rab GTPases, vesicle coat proteins and vesicle tethers.Citation2,Citation3 While SNAREs were originally postulated to impart specificity in vesicle transport,Citation4 it is now accepted that all of the other aforementioned factors can function upstream of the SNAREs and thus participate in specificity.
Upon arrival at a target membrane, the vesicle is recognized by any one of a number of possible tethering factors. These tethering factors have been classified as either coiled-coil proteins or large multisubunit tethering complexes (MTC).Citation5,Citation6 The tethering event leaves open many questions such as which tethering factor acts first or if they act simultaneously, and whether the mechanism of tethering is conserved at the various compartments. Nonetheless, it has been suggested that the tethering factors are among the first contacts between the vesicle and the target membrane and, as such, may contribute to the exquisite specificity in vesicle transport.Citation6
One of the earliest MTCs identified was the TRAPP complex.Citation7,Citation8 Although this complex has not been demonstrated to tether two distinct membranes together, it is believed to accomplish this either directly or by leading to recruitment of the factor(s) that does this. In the yeast S. cerevisiae there are three forms of the complex called TRAPP I, II and III.Citation7,Citation9-Citation11 As such, TRAPP poses a unique set of problems with respect to the role of these complexes in determining specificity in this organism. All three complexes contain the same “core” of six polypeptides (Bet3p, Bet5p, Trs20p, Trs23p, Trs31p and Trs33p) with TRAPP II containing four additional subunits (Tca17p, Trs65p, Trs120p and Trs130p) and TRAPP III containing one additional subunit (Trs85p). While TRAPP I has been implicated in ER-to-Golgi transport,Citation7,Citation11 TRAPP II has been shown to function at a later Golgi compartmentCitation7,Citation9 and TRAPP III was suggested to function in autophagy.Citation10,Citation12,Citation13 Confounding the problem of TRAPP-mediated specificity is the fact that Bet3p, a common subunit found in two copies within the core,Citation14-Citation16 was shown to bind to Sec23p, a component of the ER-derived COP II coat,Citation17 suggesting that the Sec23p-binding site on all copies of Bet3p must be blocked in both TRAPP II and III. In addition, this core of the complex was shown to be sufficient for Ypt1-directed guanine nucleotide exchange factor (GEF) activityCitation15 and indeed all three complexes have been reported to be capable of such activity.Citation7,Citation10,Citation11,Citation18,Citation19 Finally, to date only one complex has been reported in mammalian cells and it was recently shown that this complex contains homologs of the S. cerevisiae TRAPP II- and III-specific proteins.Citation20-Citation22
In an effort to better understand the relationship between the TRAPP complexes, we focused on Trs23p that links two Bet3p-containing subcomplexes to form the TRAPP holocomplex,Citation14,Citation15 thus providing GEF activity. In addition, Trs23p interacts with the GTPase Ypt1p.Citation14 Also unique to this subunit is a PDZ-like domain in higher eukaryotes and a Saccharomycotina-specific domain seen in the S. cerevisiae protein.Citation14,Citation15,Citation23,Citation24 Furthermore, co-expression of the core proteins leads to assembly of a functional complex when using the S. cerevisiae proteins but the mammalian proteins fail to assemble,Citation15 suggesting differences in the assembly and/or stability of the core between both organisms.
Using random and targeted mutagenesis we have constructed a series of mutations in Trs23p. We show that neither the C-terminal 99 amino acids (trs23Δ99C) nor the Saccharomycotina-specific domain (trs23ΔSMS) of Trs23p are essential for viability. A more extensive characterization of trs23ΔSMS showed no growth defect and that TRAPP I subunits are shifted to a much smaller molecular size suggesting that this domain is responsible for the appearance of TRAPP I in vitro. The absence of TRAPP I did not affect ER-to-Golgi traffic either in vivo or in vitro, nor later Golgi traffic or autophagy. We demonstrate that under physiological conditions TRAPP II co-fractionates with TRAPP III, and this is the only source of TRAPP seen in trs23ΔSMS. Importantly, we demonstrate that the fraction containing a mixture of TRAPP II and III can be induced to produce TRAPP I and dimeric TRAPP II by incubation in high salt. Finally, we show that in contrast to S. cerevisiae, the closely-related yeast Pichia pastoris, which lacks the SMS domain in Trs23p, does not contain a TRAPP I peak. Our data suggest that the SMS domain stabilizes the TRAPP I core complex and that this complex may be an in vitro anomaly. The implications of these findings on membrane trafficking are discussed.
Results
Neither the C-terminal 99 residues nor the SMS domain of Trs23p are essential
In an effort to examine the role of the Trs23p subunit in TRAPP function, we conducted a random mutagenesis screen using hydroxylamine to identify conditional mutations in TRS23. This screen identified a mildly temperature (heat)-sensitive (ts) and cold sensitive (cs) mutant. Upon sequencing of the mutation, TRS23 was found to contain a C361T mutation that introduced a nonsense mutation following the 120th amino acid (herein referred to as trs23Δ99C), indicating that the C-terminal 99 amino acids of Trs23p are not essential for vegetative growth of the yeast (). Further C-terminal truncations generated by either random (trs23Δ167C, trs23Δ202C) or site-directed (trs23Δ109C) mutagenesis were found to be lethal (). Alignment of S. cerevisiae Trs23p with its human counterpart (TRAPPC4) indicates that residues 56–115 comprise a domain not found in higher eukaryotes (; also see ref. Citation15). This domain is only present in species within the Saccharomycotina phylum and, hence, we will refer to it as the Saccharomycotina-specific (SMS) domain. Since truncation of Trs23p that removed even a small portion of this domain (trs23Δ109C) resulted in lethality, we tested whether the SMS domain itself was essential for function by deleting this domain (herein referred to as trs23ΔSMS). This mutant did not display a discernible growth phenotype at either extreme temperature tested () suggesting it is not essential for function. Furthermore, truncations of Trs23p from the N-terminus (removal of the first 15 amino acids) resulted in lethality (). Collectively, our data suggest that the carboxy-terminus of Trs23p does not provide an essential function to the protein and that the SMS domain is required for some aspect of Trs23p function that can be compensated by the carboxy-terminus of the protein.
Figure 1. Growth properties of trs23 mutations. (A) MSY62 was transformed with a plasmid (pRS315) containing TRS23 that was subjected to either random mutagenesis yielding nonsense mutations after the 17th, 52nd and 120th amino acids (trs23Δ202C, trs23Δ167C and trs23Δ99C, respectively); or site-directed mutagenesis to generate a nonsense mutation after amino acid 110 (trs23Δ109C) or deletion of either the first 15 amino acids (trs23Δ15N) or the SMS domain (trs23ΔSMS). Cells were plated on either SD-leucine or on 5-FOA to counterselect against wild-type TRS23 in the URA3-containing plasmid pRS316 and incubated at 30°C. (B) Serial 10-fold dilutions of wild-type or yeast strains with the mutations indicated were plated on YPD and incubated at either permissive temperature (30°C) or the restrictive temperatures of 16°C or 38°C. (C) A schematic showing all of the TRS23 mutations used in this study with their viability and growth phenotypes indicated (cs, cold sensitive; ts, heat sensitive). (D) Human TRAPPC4 (accession number NP_057230.1) and S. cerevisiae Trs23p (accession number NP_101532.1) were aligned using Clustal W and the gaps were inserted manually in accordance with Kim et al., 2006. Identities are shaded in black, the PDZ-like domain of human TRAPPC4 is underlined and the SMS domain of Trs23p is shaded in gray.
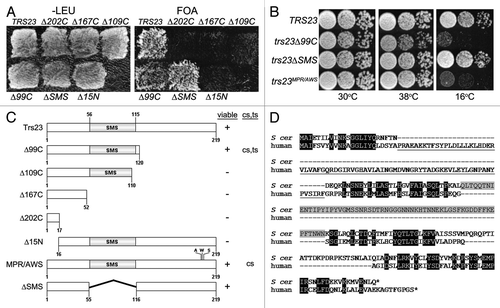
An earlier report suggested that Trs23 containing the triple point mutation M200A/P201W/R203S (herein referred to as trs23MPR/AWS) was lethal.Citation14 Since we have just demonstrated that the carboxy-terminus of Trs23p, which contains the aforementioned residues, is not essential for growth, we re-examined the growth characteristics of this triple point mutant. Consistent with our trs23Δ99C mutant, we found that trs23MPR/AWS was not lethal but did in fact display a severe cs phenotype (). We further found that trs23MPR/AWS is not dominant since neither a heterozygous diploid strain nor a haploid strain containing trs23MPR/AWS and wild-type TRS23 displayed any growth defects. Although the reason for the discrepancy in the lethality of trs23MPR/AWS is not clear, the phenotype we now show is consistent with the non-essential nature of the carboxy-terminus of TRS23.
The SMS domain is required for efficient assembly of recombinant TRAPP I
To begin to address the consequences of the removal of either the carboxy-terminus or the SMS domain of Trs23p, we expressed these proteins in our recombinant bacterial system that generates a functional TRAPP I complex (rTRAPP I) that can be detected using size exclusion chromatography.Citation15 As shown in , although the truncated trs23Δ99C protein was produced and could be detected by anti-Trs23p antibody in the cell lysate, it was largely insoluble and did not co-purify with His-tagged Bet3p. In contrast to the cells expressing wild-type Trs23p where rTRAPP I is found in a high molecular size fraction, rTRAPP I was absent in lysates expressing either the trs23Δ99C or the trs23ΔSMS proteins (). Instead, cells expressing trs23Δ99C contained a Bet3p-Trs33p-Bet5p subcomplex while those expressing trs23ΔSMS contained a Bet3p-Trs33p-Bet5p-trs23ΔSMS subcomplex.
Figure 2. Assembly and function of recombinant TRAPP containing the trs23Δ99C and trs23ΔSMS proteins. (A) Bacterial cells expressing wild-type TRAPP I proteins (with His-tagged Bet3p and Trs33p) and Trs23p (lanes 1–3) or trs23Δ99C (lanes 4–6) were probed for Trs23 using anti-Trs23p antiserum either in the total cell lysate (TCL; lanes 1 and 4), the soluble fraction (lanes 2 and 5) or in the eluate following a Ni2+-NTA agarose purification (lanes 3 and 6). (B) Bacterial cells expressing wild-type TRAPP I proteins (where Bet3p is His-tagged) and Trs23p, trs23Δ99C or trs23ΔSMS as indicated, were first purified with Ni2+-NTA agarose and then the eluates were fractionated by size exclusion chromatography on a Superdex 200 column. Fractions from the column were then analyzed by SDS-PAGE and the gel was silver stained. The positions of the TRAPP proteins are indicated to the side of each panel and the position on the column of fully-assembled recombinant TRAPP I is indicated by an asterisk (*). (C) Lysates from the bacterial cells in (B) were assayed for Ypt1p GEF activity as described in Experimental Procedures [wild-type (■); trs23Δ99C (●); trs23ΔSMS (○)]. The intrinsic ability of Ypt1p to release nucleotide is also shown (Δ). Assays represent three replicates and error bars represent ± SEM.
![Figure 2. Assembly and function of recombinant TRAPP containing the trs23Δ99C and trs23ΔSMS proteins. (A) Bacterial cells expressing wild-type TRAPP I proteins (with His-tagged Bet3p and Trs33p) and Trs23p (lanes 1–3) or trs23Δ99C (lanes 4–6) were probed for Trs23 using anti-Trs23p antiserum either in the total cell lysate (TCL; lanes 1 and 4), the soluble fraction (lanes 2 and 5) or in the eluate following a Ni2+-NTA agarose purification (lanes 3 and 6). (B) Bacterial cells expressing wild-type TRAPP I proteins (where Bet3p is His-tagged) and Trs23p, trs23Δ99C or trs23ΔSMS as indicated, were first purified with Ni2+-NTA agarose and then the eluates were fractionated by size exclusion chromatography on a Superdex 200 column. Fractions from the column were then analyzed by SDS-PAGE and the gel was silver stained. The positions of the TRAPP proteins are indicated to the side of each panel and the position on the column of fully-assembled recombinant TRAPP I is indicated by an asterisk (*). (C) Lysates from the bacterial cells in (B) were assayed for Ypt1p GEF activity as described in Experimental Procedures [wild-type (■); trs23Δ99C (●); trs23ΔSMS (○)]. The intrinsic ability of Ypt1p to release nucleotide is also shown (Δ). Assays represent three replicates and error bars represent ± SEM.](/cms/asset/3ba20e50-1cfd-4d71-a79c-f24d759e11a1/kcll_a_10919414_f0002.gif)
Since small amounts of rTRAPP I may be present in the lysates generated from the two mutants, we assayed the lysate for guanine nucleotide exchange factor (GEF) activity for the GTPase Ypt1p since this activity is robust with rTRAPP I.Citation14,Citation15 Compared to lysate containing wild-type Trs23p and consistent with no detectable rTRAPP I, lysate with trs23Δ99C displayed near background levels of GEF activity (). In contrast, lysate prepared from cells expressing the trs23ΔSMS protein displayed GEF activity intermediate to that of wild-type and Ypt1p alone () suggesting that small, but undetectable, amounts of rTRAPP I do indeed form in these cells. Our results suggest that, in vitro, rTRAPP I cannot assemble with the trs23Δ99C protein. They further suggest that the SMS domain of Trs23p is not essential for GEF activity of rTRAPP I but is required for its efficient assembly.
The SMS domain contributes to Trs23p interactions
The lack of efficient assembly of rTRAPP I in the presence of either the trs23ΔSMS or trs23Δ99C proteins suggests that interactions with other TRAPP subunits are abrogated. To test this notion, wild-type Trs23p, trs23ΔSMS and trs23Δ99C were expressed as fusion proteins with the Gal4p DNA binding domain. The plasmid expressing these proteins was introduced into yeast containing open reading frames encoding individual TRAPP proteins fused to the Gal4 transcriptional activation domain. Reconstitution of Gal4p-dependent transcription, and hence a protein-protein interaction, was assessed by three criteria: growth of the cells on medium (1) lacking histidine, (2) lacking both histidine and adenine and (3) lacking histidine but containing 3-aminotriazole (3AT), a competitive inhibitor of His3p. Wild-type Trs23p displayed interactions with Trs31p, Bet5p and Trs20p (). Interactions with Trs31p and Bet5p were also seen in the crystal structure of both the yeast and mammalian proteins.Citation14,Citation15 While an interaction with Trs20p was not detected in the crystal structure, a similar interaction between the mammalian homologs TRAPPC4 and TRAPPC2 was recently reportedCitation21 and may reflect interactions in higher ordered structures.Citation25 The trs23Δ99C protein lost all of the interactions seen with the wild-type protein, consistent with its inability to support assembly of rTRAPP I. The trs23ΔSMS protein lost its ability to interact with Bet5p and showed a weakened ability to interact with both Trs31p and Trs20p (), suggesting that the SMS domain contributes to these interactions. These differences were not due to reduced expression levels or instability of the mutant proteins (). Weakened interactions between trs23ΔSMS and other TRAPP proteins helps to explain the intermediate GEF activity seen in bacterial lysates containing trs23ΔSMS () in the absence of detectable amounts of rTRAPP I ().
Figure 3. The SMS domain of Trs23p contributes to the strength of Trs23p-TRAPP subunit interactions. (A) Wild-type Trs23p or the mutants trs23Δ99C and trs23ΔSMS were expressed in yeast as fusion proteins with the DNA binding domain of Gal4p and mated to yeast strains expressing other TRAPP subunits fused to the transcriptional activation domain of Gal4p. Serial 10-fold dilutions of diploids were plated on SD-leucine/tryptophan, SD-leucine/tryptophan/histidine, SD-leucine/tryptophan/adenine and SD-leucine/tryptophan/histidine containing 5mM 3AT and all plates were incubated at 30°C. A two-dimensional schematic of the TRAPP II complex is shown to the right of the panel. Shaded in gray is the TRAPP I core of the complex whose crystal structures and organization are known. The position and interactions of the remaining subunits (stippled) are not known but are based on previously published studies on the organization of yeast and mammalian TRAPP complexes.Citation25,Citation27,Citation36 (B) Lysates were produced from yeast cells expressing Trs23p, or the mutant proteins trs23ΔSMS or trs23Δ99C from the plasmid pGBKT7. Equal amounts of protein were loaded in each lane and probed with anti-myc IgG since the fusion proteins contain a myc epitope tag. Identical results were obtained when the blot was probed with anti-Trs23p IgG (not shown).

trs23ΔSMS retains Ypt1p GEF activity in yeast lysates
We next explored the level of Ypt1p GEF activity in the yeast mutants trs23ΔSMS and trs23Δ99C. In lysates prepared from trs23ΔSMS, GEF activity was comparable to that of wild-type () confirming that this domain is not essential for GEF activity. In contrast, lysates from trs23Δ99C were reduced to near background levels and was similar to that of trs23MPR/AWS () that was previously shown to be defective in vitro (see ref. Citation14). These results are similar to those seen in vitro () and suggest that the assembly of TRAPP in trs23ΔSMS is more efficient in vivo compared with in vitro. Furthermore, the stability of TRAPP appears to be largely unaffected in trs23ΔSMS, in contrast to trs23Δ99C where several subunits are less stable (). Reduced levels of TRAPP could account for the growth phenotype seen for trs23Δ99C. For these reasons, we focused our attention on the trs23ΔSMS mutant which, although has weakened interactions, displays no defects in growth, TRAPP subunit levels or GEF activity in yeast.
Figure 4. GEF activity is unaffected in S. cerevisiae containing trs23ΔSMS. (A) Lysates from yeast cells expressing wild-type Trs23p (■) or the mutant proteins trs23Δ99C (●), trs23ΔSMS (○) or trs23MPR/AWS (X) were prepared and assayed for Ypt1p-directed GEF activity as described in Experimental Procedures. The intrinsic ability of Ypt1p to release nucleotide is also shown (▾). (B) Lysate from wild-type (■) or trs23ΔSMS (○) was fractionated by size exclusion chromatography on a Superose 6 column in 150mM salt. The fractions enriched in TRAPP II/III (fractions 16–17) and TRAPP I (fractions 29–31) were pooled, concentrated and assayed for Ypt1p GEF activity compared with the intrinsic ability of Ypt1p to release nucleotide. The intrinsic ability of Ypt1p to release nucleotide is also shown (▾). Assays in (A) and (B) represent three replicates and error bars represent ± SEM.
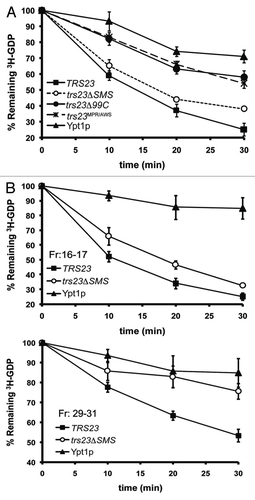
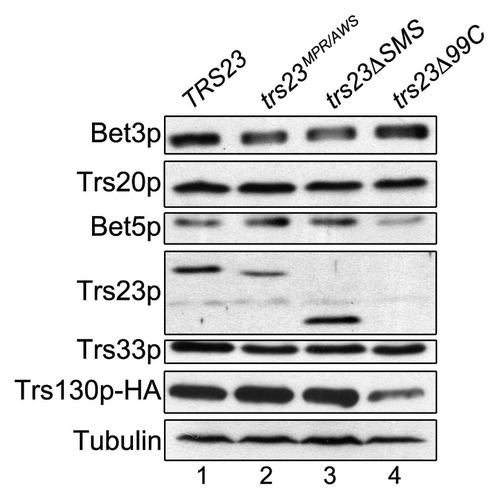
Figure 5. The levels of TRAPP subunits are largely unaffected in trs23ΔSMS. Lysates from wild-type (lane 1), trs23MPR/AWS (lane 2), trs23ΔSMS (lane 3) and trs23Δ99C (lane 4) were probed with antibodies recognizing the TRAPP subunits indicated or anti-HA to detect endogenously-tagged Trs130p. Tubulin served as a loading control.
trs23ΔSMS contain only a single TRAPP peak at physiological salt
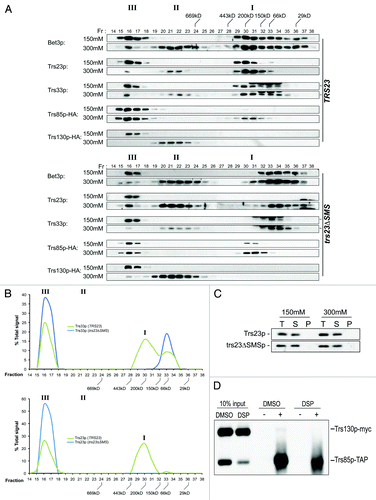
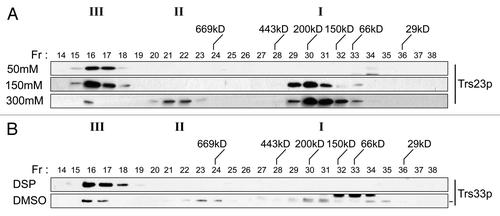
Our results thus far suggest that the trs23ΔSMS protein has reduced interactions with TRAPP proteins and only weakly supports the formation of the TRAPP core in vitro yet yeast bearing this mutation are not defective in GEF activity. We therefore explored the effects of this mutation on the formation of the three reported yeast TRAPP complexes TRAPP I, II and III. Lysates from wild-type and trs23ΔSMS were fractionated in physiological salt (150 mM) by size exclusion chromatography on a Superose 6 column. This column was chosen since, unlike Superdex 200 that has been used for most previously-reported TRAPP studies, it readily separates TRAPP II and III.Citation20 Consistent with previous studies,Citation7,Citation11,Citation18,Citation20,Citation26,Citation27 Bet3p, Trs23p and Trs33p in wild-type lysates are found in two high molecular weight fractions (). While these have previously been reported to correspond to TRAPP II and I on a Superdex 200 column, we now show the remarkable result that the highest molecular weight peak (fractions 16–17) contains both the TRAPP III- and II-specific subunits Trs85p and Trs130p, respectively (). Only at higher salt concentrations (300 mM) do we see these proteins separate into two peaks largely distinct from each other which are indicated as TRAPP III (fractions 16–17) and II (fractions 21–22) (). The third TRAPP complex, TRAPP I, peaks at fractions 30–31 (). In lysates from trs23ΔSMS, similar results were seen with respect to the peak seen in fractions 16–17, and the salt-induced separation into distinct TRAPP III and II peaks was also noted (). However, in trs23ΔSMS we no longer detected the TRAPP I peak that appears in fractions 30–31 in wild-type cells. Rather, the TRAPP subunits Bet3p, Trs23p and Trs33p are found in a peak in fractions 33/34 (). This latter peak has a molecular size of ~67 kD which likely corresponds to monomers or very small subcomplexes of these subunits. In addition, we noted the appearance of a sizeable pool of Trs85p in trs23ΔSMS that increases under high salt conditions. The complexes seen on this column are likely representative of the cellular pool of TRAPP since the majority of TRAPP was loaded onto the columns at both 150 mM and 300 mM salt (). Collectively, our results demonstrate that, at physiological salt, trs23ΔSMS contain just a single TRAPP peak corresponding to the region where TRAPP III fractionates and that under these conditions the TRAPP II peak is not present even in wild-type.
Figure 6. A single TRAPP peak is detected in trs23ΔSMS at physiological salt concentrations. (A) Lysates from wild-type and trs23ΔSMS were fractionated on a Superose 6 size exclusion column in buffer containing either 150 mM NaCl or 300 mM NaCl as indicated. Fractions of 0.5 ml were collected and probed with antibodies that recognize Bet3p, Trs33p, Trs23p or HA (to detect endogenously-tagged Trs130p or Trs85p). I, II and III above the wild-type and trs23ΔSMS blots indicate the location of TRAPP I, II and III, respectively, under conditions where they separate from each other. Molecular size standards are also indicated above the blots. Note that a cross-reactive band appears just above the Trs33p band in fractions 32–34 with the anti-Trs33p antibody and the dash to the right of the Trs33 panels indicates the position of Trs33p. For each subunit shown, samples ranging from fractions 14–38 were fractionated on two separate polyacrylamide gels and processed for western analysis simultaneously. Exposures for each half of the two gels were identical. (B) The signals for Trs33p (top panel) and Trs23p (bottom panel) from wild-type (green) and trs23ΔSMS (blue) at 150 mM NaCl shown in (A) were quantitated using Image J and plotted as a percentage of the total signal. Note the absence of the TRAPP I peak in trs23ΔSMS. (C) Lysates were prepared for fractionation as in (A) at either 150 mM or 300 mM NaCl. Samples before the centrifugation (T), the pellet fraction following centrifugation (P) and the supernatant (S) that was loaded onto the Superose 6 column were probed for Trs23p in both wild-type and trs23ΔSMS. (D) Lysate from wild-type cells containing Trs85p-TAP and Trs130p-myc were fractionated on a Superose 6 column and fractions 16–17 were collected and split in two. One sample was treated with DMSO while the other sample was treated with the crosslinking reagent DSP for 3 h on ice. Each sample was then split in two and either treated with (+) or without (-) anti-TAP IgG. The immune complexes were collected on protein A-sepharose beads, fractionated by SDS-PAGE and probed with anti-myc IgG. Samples representing 10% of the input are shown.
The fact that Trs85p and Trs130p co-fractionated in a single peak at physiological salt in both wild-type and trs23ΔSMS led us to consider the possibility that there is a single TRAPP complex that is unstable and easily fragments with increasing salt. Alternatively, these two TRAPP complexes could be either oligomerized or bound to other cellular components causing them to co-fractionate. To distinguish between these possibilities we constructed a strain that contains TAP-tagged Trs85p and Trs130p-myc. The strain was fractionated under conditions where only the TRAPP III peak, but not the TRAPP II peak, was detected (50 mM NaCl; see ). The TRAPP III-containing fraction was then treated with the crosslinking reagent DSP or left untreated before processing for immunoprecipitation. Although crosslinking was efficient (see ), when Trs85p was immunoprecipitated with anti-TAP IgG we failed to detect co-precipitation of Trs130p-myc even though > 50% of the cellular Trs85p was immunoprecipitated (). Since multiple copies of Trs85p were not previously detected in TRAPP IIICitation20 and since at least two copies of Trs130p are present in TRAPP II,Citation25 we should have easily been able to detect co-precipitation of Trs130p-myc if there was just a single complex in fractions 16–17. Therefore, we conclude that at physiological salt, TRAPP II and III represent distinct complexes that co-fractionate on a Superose 6 column.
Figure 8. TRAPP I can form from TRAPP II/III. (A) Lysates of wild-type yeast were prepared in buffer containing 50 mM, 150 mM or 300 mM NaCl and fractionated by size exclusion chromatography on a Superose 6 column in buffer containing identical salt. The fractions were analyzed by western blotting for the presence of Trs23p. (B) The TRAPP II/III-containing fraction from wild-type lysate that was fractionated in the presence of 50 mM NaCl was incubated with 2 mM DSP or DMSO, quenched with Tris pH 7.5 and re-fractionated on a Superose 6 column in buffer containing 300 mM NaCl. Fractions were probed for the presence of Trs33p. Note that a cross-reactive band appears just above the Trs33p band in fractions 32–34 with the anti-Trs33p antibody in the DMSO-treated sample and the dash to the right of the Trs33 panel indicates the position of Trs33p. I, II and III above the wild-type and trs23ΔSMS blots indicate the location of TRAPP I, II and III, respectively, under conditions where they separate from each other.
TRAPP II/III provides Ypt1p GEF activity in trs23ΔSMS
The ability of TRAPP I to act as a GEF for Ypt1p is firmly established.Citation7,Citation14,Citation15,Citation18,Citation28 The absence of TRAPP I in trs23ΔSMS along with near wild-type levels of GEF activity in vivo, suggested that an alternate Ypt1p GEF was functioning to support ER-to-Golgi transport. One possibility is that Dss4p, a protein that facilitates nucleotide release from several GTPases including Ypt1pCitation29 and genetically interacts with TRAPP,Citation30 can facilitate nucleotide exchange on Ypt1p in the absence of TRAPP I. If this were the case, we would expect to see a genetic interaction between DSS4 and TRS23. Since trs23Δ99C has an obvious growth phenotype, we used this mutant to explore genetic interactions with DSS4. Overexpression of DSS4 did not alleviate either the ts or cs phenotype of trs23Δ99C (not shown). In addition, tetrad analysis following a cross between the two haploid mutant yeast strains dss4Δ and trs23Δ99C did not reveal any growth defect or exacerbation of the growth phenotype seen for trs23Δ99C (22 tetrads dissected, co-segregation in 19 colonies). These results argue against Dss4p compensating for the lack of TRAPP I.
Alternatively, TRAPP II or III, the only TRAPP complexes detected in trs23ΔSMS at physiological salt, may act as a Ypt1p GEF since previous literature in this area in which lysates were assayed for GEF activity has been controversial.Citation7,Citation14,Citation18,Citation28 Yeast lysates from wild-type and trs23ΔSMS were fractionated by size exclusion chromatography on a Superose 6 column and fractions 16–17 (TRAPP II/III) and 29–31 (where TRAPP I fractionates) were assayed for Ypt1p GEF activity. Indeed, both the TRAPP II/III- and TRAPP I-containing fractions displayed Ypt1p GEF activity in wild-type. In contrast, and consistent with the absence of TRAPP I in trs23ΔSMS, this mutant only displayed Ypt1p GEF activity in the TRAPP II/III fractions (). It is noteworthy that although TRAPP III has been reported to display Ypt1p GEF activity,Citation10 the Trs85p peak near TRAPP I is not active in our assay, suggesting that only the high molecular weight peak of Trs85p possesses this activity. Our results suggest that in the absence of TRAPP I, either TRAPP II or III can provide sufficient Ypt1p GEF activity to maintain cell growth.
trs23ΔSMS does not display defects in membrane traffic
The only notable defect seen in trs23ΔSMS has been the disappearance of its TRAPP I peak. Since TRAPP I is reported to function in ER-to-Golgi transport, we expected that a defect in this stage of membrane traffic would manifest in a growth phenotype. However, this was not seen (see ). In order to more closely assess this mutant for possible defects in membrane traffic we first examined the processing of the vacuolar protease carboxypeptidase Y (CPY) by pulse-chase analysis. This protein has ER (p1), Golgi (p2) and vacuolar (m) forms that are easily distinguished by SDS-PAGE. As shown in , trs23ΔSMS did not show a noticeable defect in CPY processing. The pattern was similar to wild-type and stood in stark contrast to that of sec18, which displayed the expected block in ER-to-Golgi traffic as evidenced by the failure of the cells to process p1CPY.
Figure 7. Membrane trafficking is unaffected in the trs23ΔSMS mutant. (A) Wild-type, trs23ΔSMS and sec18 yeast were pulse-labeled with 35S-methionine/cysteine and chased for the times indicated below the panels. Carboxypeptidase Y (CPY) was immunoprecipitated from lysates at each time point, fractionated by SDS-PAGE and visualized by autoradiography. The position of the ER (p1) and vacuolar (m) forms of CPY are indicated. (B) Wild-type, trs23ΔSMS and trs85Δ were transformed with a plasmid expressing GFP-Snc1p. The cells were fixed and viewed using an epifluorescence microscope. (C) Wild-type, trs23ΔSMS and trs23Δ99C were transformed with a plasmid expressing Ypt31p-GFP. The cells were fixed and viewed using an epifluorescence microscope. For quantitation in (B) and (C), a minimum of 100 cells from three replicates were counted. The error bars represent ± SEM and representative cells used for quantitation are shown as insets. (D) Wild-type, trs23ΔSMS and trs85Δ were transformed with a plasmid expressing Ape1p-GFP, grown in minimal medium without uracil, converted to spheroplasts and lysed. Samples were fractionated by SDS-PAGE and probed with anti-GFP. The percentage of Ape1p-GFP processing, calculated using Image J, is indicated below each lane. (E) Reconstitution of ER-to-Golgi traffic was performed as described in Experimental Procedures. Reactions were performed with no additions either on ice (ice) or at 29°C (NA), or fully reconstituted at 29°C (Recon). The results are expressed as percentage of concanavalin A-precipitable pro-α-factor that has received α-1,6-mannose Golgi modifications. The results are the average of duplicates and the range of transport over three independent experiments was 13.6–15.4% (wild-type) and 12.6–14% (trs23ΔSMS). (F) Lysates were prepared from wild-type and trs23ΔSMS in buffer B88 that was used for the in vitro transport assay in (E) and fractioned on a Superose 6 column in B88. Fractions were probed with anti-Trs33p. I, II and III above the blots indicate the location of TRAPP I, II and III, respectively, under conditions where they separate from each other.
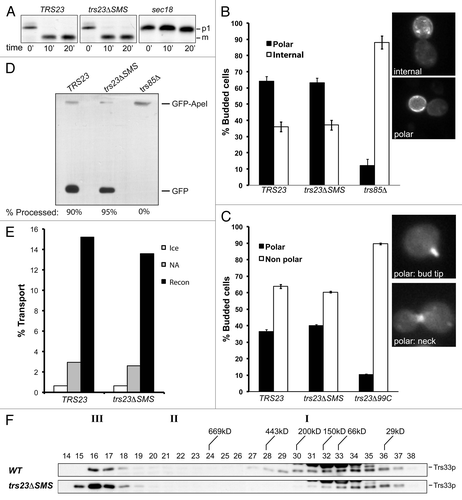
Since Trs23p is a component of TRAPP II that reportedly functions at the late Golgi, we also examined the cells for defects in later stages of the secretory pathway. Snc1p, a protein that cycles between the late Golgi and the plasma membrane, is normally found at steady-state on the plasma membrane of unbudded yeast cells or on the emerging bud in a polarized manner when fused to GFP (GFP-Snc1p).Citation31 Defects in recycling lead to the accumulation of Snc1p inside of the cells or a loss of its polarized distribution. Wild-type cells displayed the expected distribution of GFP-Snc1p (). As was the case with CPY processing, GFP-Snc1p localization was unaffected in trs23ΔSMS. This result contrasts with that of trs85Δ which showed a strong GFP-Snc1p recycling defect () as previously reported.Citation27
To further explore trs23ΔSMS for a late secretory pathway defect we examined the localization of the fluorescently-tagged GTPase GFP-Ypt31p. This GTPase normally concentrates in small buds and at the mother/bud neck,Citation32 which we saw in wild-type cells (). This pattern was unaffected in trs23ΔSMS, in contrast to trs23Δ99C where it was clearly altered ().
TRAPP III has recently been reported to function in autophagy.Citation10 We therefore examined the cytosol-to-vacuole targeting (cvt) pathway in trs23ΔSMS using the marker Ape1p-GFP. This protein is processed by the cvt pathway and is ultimately cleaved releasing GFP which is detected by western analysis.Citation33 As shown in , both wild-type and trs23ΔSMS were capable of processing Ape1p-GFP. In contrast, and as previously reported,Citation12,Citation13 trs85Δ cells were defective in this pathway ().
We also examined trs23ΔSMS for defects in an in vitro assay that reconstitutes ER-to-Golgi transport.Citation34 This assay measures the acquisition of Golgi-specific modifications of a marker protein called pro-α-factor. Compared with wild-type, trs23ΔSMS did not show a defect in transport in vitro (). As was shown earlier using physiological salt (see ), lysates from trs23ΔSMS prepared in the buffer used for the in vitro assay (buffer B88) also showed a single TRAPP peak corresponding to TRAPP II/III (). Interestingly, lysates from wild-type used for this assay showed a broadening of the TRAPP I peak suggesting this complex is not stable in this buffer. Collectively, our results suggest that trs23ΔSMS does not adversely affect membrane traffic in vitro nor in vivo.
TRAPP I can form from TRAPP II/III in vitro
Our results thus far suggest that in the absence of TRAPP I, cells survive with no adverse effects in ER-to-Golgi traffic. This was surprising given that TRAPP I is implicated in this pathway. One explanation is that TRAPP I represents either an intermediate in the assembly of TRAPP II and/or III or is a fragment of either complex since we found that its appearance is salt-dependent (). To address this we first fractionated a lysate from wild-type cells in low (50 mM) salt where only the TRAPP II/III peak is detected (). We then incubated this fraction either with or without the crosslinking reagent DSP and fractionated the sample in 300 mM salt, conditions where all three TRAPP complexes are detected (see ). As shown in , while the untreated sample did indeed show a TRAPP I peak, the crosslinked sample did not. Importantly, no intermediate peak between TRAPP II/III and TRAPP I was detected in the untreated sample. These results suggest that TRAPP I is a subcomplex of one or both of the larger (TRAPP II and III) complexes that forms in the presence of increasing salt in vitro.
TRAPP I appearance is SMS domain-dependent
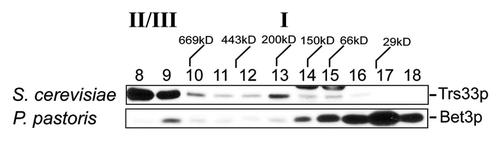
TRAPP I, as defined by the co-fractionation of the TRAPP core subunits on a gel filtration column, is readily seen in S. cerevisiae but not in human cells.Citation21,Citation22,Citation35,Citation36 We speculated that this may be due to added stability afforded to this complex by the SMS domain of Trs23p. Like the human protein, Trs23p in other yeast such as P. pastoris do not have the SMS domain. Thus, if our hypothesis is correct, we would expect that P. pastoris would not display a TRAPP I peak. We found that our antibody raised against the human TRAPPC3 protein, the most conserved subunit in TRAPP, was cross-reactive with P. pastoris Bet3p providing us with a means to address whether this yeast has a TRAPP I peak. Lysate from P. pastoris and S. cerevisiae was fractionated by size exclusion chromatography on a Superdex 200 column to better resolve the smaller molecular size region where TRAPP I fractionates. In S. cerevisiae, TRAPP I is detected in fraction 13 while TRAPP II/III is detected in fractions 8–9 (). In the P. pastoris lysate, a high molecular weight pool of Bet3p was detected that most likely corresponds to TRAPP II/III (). Based on the size of the P. pastoris TRAPP subunits, a TRAPP I complex would be expected to be only 30 kD smaller than the S. cerevisiae complex. While a second peak of Bet3p was detected, it eluted in a fraction (fraction 17) with a molecular size much smaller than that expected for TRAPP I and no other peak of P. pastoris Bet3p was seen. The absence of a TRAPP I-equivalent was also noted for human cells, whose Trs23p homolog does not contain an SMS domain.Citation21,Citation22,Citation36,Citation37 This result is consistent with the notion that the SMS domain of Trs23p stabilizes S. cerevisiae TRAPP I.
Figure 9.P. pastoris does not contain a TRAPP I peak. Lysates in 150 mM NaCl from P. pastoris and S. cerevisiae were fractionated by size exclusion chromatography on a Superdex 200 column. The fractions were probed for Trs33p (S. cerevisiae) or P. pastoris Bet3p using anti-TRAPPC3 IgG. The positions of TRAPP II/III and I are indicated above the top panel.
Discussion
Here we demonstrate that the Saccharomycotina-specific (SMS) domain of S. cerevisiae Trs23p is not required for Ypt1p-directed GEF activity but is important for the stable interaction between Trs23p and other TRAPP subunits. Our data suggest that cells devoid of this domain of Trs23p contain two large TRAPP complexes called TRAPP II and III and do not contain the smaller TRAPP I complex. These cells resemble wild-type with respect to four different membrane trafficking assays both in vivo and in vitro and in terms of their growth characteristics. Based on our study, we suggest that the SMS domain of Trs23p stabilizes fragments (TRAPP I) of TRAPP II and/or III.
Collectively, our data may suggest that S. cerevisiae contain a single Golgi-localized TRAPP complex (TRAPP II) that is capable of supporting ER-to-Golgi traffic. This is based on a number of observations including: (1) removal of the SMS domain of Trs23p in S. cerevisiae results in a loss of the TRAPP I peak with no corresponding reduction in Ypt1p GEF activity or noticeable growth phenotype; (2) ER-to-Golgi traffic is normal both in vivo and in vitro; (3) a TRAPP I peak can be formed by incubating the TRAPP II/III peak with high salt in vitro. Since TRAPP II, but not TRAPP III, localizes to the Golgi we propose that TRAPP II can function in ER-to-Golgi transport. Since we cannot separate TRAPP II from III, we cannot rule out the possibility that TRAPP III is the complex that functions in ER-to-Golgi traffic. In this respect it is noteworthy that depletion of Trs85p, the TRAPP III-specific subunit, was reported to be defective in ER-to-Golgi trafficCitation7 and autophagy may use membranes derived from the ERCitation38 suggesting the possibility that the role of TRAPP III in autophagy may be an indirect result of its role in ER-to-Golgi traffic.
An important next step will be to characterize the TRAPP II and III superstructures that we report. We have shown an identical fractionation pattern to that seen in in the presence of Triton X-100 for the TRAPP II subunit Trs130p (S.B. and M.S., unpublished observation), suggesting that these structures are either bound to detergent-insoluble membranes, they oligomerize to greater than a dimer or are bound to a large protein structure as previously suggested.Citation16 In addition, uncharacterized subunits may be present in the complex. An alternative explanation for our data are that in the trs23ΔSMS cells, TRAPP I is more tightly associated with the TRAPP superstructures and would require higher salt to be liberated. We do not favor this possibility since increasing the salt concentration in trs23ΔSMS redistributes TRAPP core proteins from the higher molecular size fractions to a smaller fraction. The resulting peak is significantly smaller than TRAPP I and, at physiological salt, is devoid of Trs23p. It is evident that data addressing the existence and function of TRAPP I (which has only been demonstrated in vitro) as an independent TRAPP complex must be cautiously interpreted in light of our in vitro data.
Our results implicate the SMS domain of Trs23p in the stability of TRAPP I. The added stability afforded to TRAPP I by the SMS domain may explain why a recombinant form of human TRAPP I could not be assembled like that seen for the S. cerevisiae subunits.Citation15 Although it remains a formal possibility that small amounts of TRAPP I can support growth with no observable phenotype, given the very low levels of TRAPP in cells to begin withCitation16 and the amounts of TRAPP I needed to support ER-to-Golgi transport in vitro,Citation7 as well as the lack of GEF activity in the TRAPP I fraction of trs23ΔSMS () we do not favor this possibility.
Consistent with the notion that TRAPP I may be a fragment of TRAPP II or III is the fact that TRAPP I is not detected in live yeast cells.Citation27 In addition, it has previously been shown that the relative distribution of core subunits between the two TRAPP peaks on a Superdex 200 size exclusion column (which does not resolve TRAPP II and III) is highly variable but TRAPP II/III levels of these subunits are generally higher than those in TRAPP I.Citation7,Citation20,Citation26,Citation27 The only exceptions to this are when high salt (500 mM) is used in the preparation of the lysateCitation27 or when certain trs120 or trs130 (TRAPP II) mutants are used.Citation26 The former is consistent with the notion that TRAPP I is an in vitro, salt-dependent fragment of TRAPP II and/or III, and the latter is consistent with the role of Trs120p and Trs130p in the overall architecture of TRAPP II.Citation25 The notion that TRAPP I results from TRAPP II/III is analogous to a recent study that suggested that there were two Trs85p peaks on a size exclusion column (confirmed in the present study), where the low molecular weight peak may represent a breakdown product or an assembly intermediate of the higher molecular weight TRAPP III peak.Citation20
If our suggestion of TRAPP I being an in vitro, salt-dependent fragment of TRAPP II/III is correct, this has strong implications for the role of tethering factors as determinants of specificity and for the mechanism of action of TRAPP in ER-to-Golgi traffic in particular. The Golgi would in fact have a single TRAPP complex (TRAPP II) that could function in traffic at both ends of the organelle. In mammalian cells the early secretory pathway is more complex with a pre-Golgi ERGIC compartmentCitation39 and the need to transport diverse-sized cargo.Citation40 Whether the mammalian TRAPP complex functions at both ends of the Golgi is still unknown. However, it has been shown to function in the early secretory pathwayCitation21,Citation22,Citation36,Citation37,Citation41 and only a single complex containing all S. cerevisiae TRAPP subunit homologs has been reported, suggesting that if mammalian TRAPP does indeed function at the late Golgi then a single complex would be expected to perform both of these functions. Indeed, parallels between S. cerevisiae and human TRAPP proteins with respect to their interactions with vesicle coat proteins have already been made.Citation9,Citation17,Citation22 We suggest that factors on the vesicles (e.g., GTPases and SNAREs) in combination with coat proteins and tethers contribute to the overall specificity of vesicle transport. This notion helps to explain why some GTPases such as Ypt1p can function at multiple steps in membrane trafficCitation10,Citation42-Citation44 and why some yeast SNAREs such as Sed5p, Ykt6p and Vti1p can also function in multiple membrane trafficking processes.Citation45-Citation48 In this respect, S. cerevisiae TRAPP II and mammalian TRAPP would be no different, with the particular combinations of factors and the combined effect of multiple layers of partial specificity dictating the overall specificity of the process.
Whether S. cerevisiae TRAPP functions on the Golgi where it interacts with incoming vesicles or binds to vesicles and interacts with a Golgi-localized protein is not clear and needs to be resolved. However, recent studies suggest the latter may be possibleCitation9,Citation17,Citation22 and the mammalian Bet3p homolog, TRAPPC3, has been shown to be required for homotypic vesicle fusion suggesting that at least this subunit can bind to vesicles independent of the Golgi.Citation41 In addition, other MTCs or parts of these complexes such as the exocyst and the COG complex have been reported to localize to vesiclesCitation49-Citation51 leaving open the question of whether MTCs bind first to vesicles or target organelles. If TRAPP were to function analogously, it would have the ability to bind to both COP I and II vesicles.
Given the large size of TRAPP II and III, and the fact that the core subunit Bet3p binds to the inner Sec23/24p layer of the COP II coat, how can we envision TRAPP II or III functioning in ER-to-Golgi transport? The dimensions of a TRAPP II dimer (225 Å × 250 Å)Citation52 may in theory have access to the Sec23/24p layer since the outer layer, composed of Sec13/31p, has openings of ~300 Å.Citation52 However, TRAPP II superstructures, such as those we presently demonstrate to occur at physiological salt, would seem to be prohibitively large to have similar access. In addition, it is unlikely that the openings presented by the COP II cage are completely unobstructed given the density and numbers of proteins that can be found on vesicles.Citation53 In the case of COP II vesicles formed in vitro, the extent to which the outer layer of the coat remains intact is unclearCitation54 and it has been suggested that either a portion of each vesicle has lost its outer layer or a portion of the vesicle population has lost its entire outer layer.Citation55 In this case, the question of access of either TRAPP II or III to the Sec23/24p layer would be mitigated. It is important to stress that the extent to which the vesicle coat remains intact in vivo is not known.
Materials and Methods
Strain construction
A list of all strains used in this study is shown in . All trs23 mutations were constructed using the same parental strain (MSY62). The trs23 mutations were generated using the splicing by overlap extension PCR method or random mutagenesis. Mutant strains were created by plasmid shuffling using 5-fluoroorotic acid (5-FOA) counterselection. Endogenous TRS130 and TRS85 were tagged at the carboxy-terminus with a hemagglutinin (HA) epitope by genomic insertion of a cassette amplified from pFA6a-3HA-HIS3MX6.Citation56 Insertion at the correct location was verified by PCR and western blot analysis.
Table 1. Yeast strains used in this study
Spotting
Yeast strains were inoculated into 3 ml of minimal media and grown in a shaker incubator overnight at 30°C. The OD600 was normalized to the lowest value and 10-fold serial dilutions were spotted onto minimal media with 5-FOA or YPD (1% yeast extract, 2% peptone, 2% glucose). Yeast were placed at the permissive temperature of 30°C to serve as a growth control and at the restrictive temperatures of 16°C and 38°C.
Preparation of yeast lysates
For fractionation of yeast cytosols, cells were grown to mid log phase (OD600 of 1–1.5) overnight at 25°C. Cells were converted to spheroplasts as previously described.Citation57 Spheroplasts were lysed in lysis buffer [20 mM HEPES pH 7.3, 2 mM EDTA, 50–300 mM NaCl, 1 mM DTT, and 1X protease inhibitor cocktail (PIC)], dounce homogenized and centrifuged at 21,000 g (high speed spin) in an SW55Ti rotor for 15 min at 4°C.
For GEF assays, membrane extraction and subcellular fractionation and examining levels of TRAPP proteins, cells were grown to early log phase (OD600 of 1–1.5) overnight at 25°C. Cells were converted to spheroplasts as above, lysed in lysis buffer (20 mM HEPES pH 7.3, 1 mM DTT, 1x PIC), dounce homogenized and centrifuged at 500 g for 5 min to remove unbroken cells and debris. The supernatant was centrifuged at 16,000 g (low speed spin) in a table-top microcentrifuge for 10 min at 4°C.
Sucrose gradient fractionation and membrane floatation
For equilibrium fractionation, the 16,000 g pellet from 400 OD600 units of starting material was resuspended in 500 μl of 5% sucrose, dounce homogenized and layered on top of a discontinuous sucrose gradient (1 ml each of 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55% and 500 μl of 60% sucrose). Gradients were centrifuged for 17 h at 178,000 g in an SW41Ti rotor at 4°C. Fractions of 500 μl were collected from top to bottom with a peristaltic pump.
For membrane floatation the 16,000 g pellet from 400 OD600 units of starting material was resuspended in 500 μl of 60% sucrose and placed on the bottom of a centrifuge tube. A discontinuous step sucrose gradient was layered on top (1 ml each of 50%, 40%, 30%, 20% and 10% sucrose) and gradients were centrifuged for 4 h at 178,000 g in an SW55Ti rotor. Fractions of 500 μl were collected from top to bottom with a peristaltic pump. Sucrose solutions (w/w) were dissolved in 20 mM HEPES pH 7.3.
Size exclusion chromatography
For comparison between wild-type and trs23ΔSMS, 5 mg of total protein from a 21,000 g supernatant was fractionated in the following gel filtration buffer: 20 mM HEPES pH 7.3, 50–300 mM NaCl, 1 mM DTT (this component was excluded in crosslinking experiments). In buffer B88 was used.Citation34
Concentrated AKTA fractions for guanine nucleotide exchange factor assays (see below) were prepared by fractionating 20 mg of total protein from a 21,000 g supernatant (10 mg at a time) and fractions were concentrated using a Millipore Amicon centrifugal filter to a final volume of 200 μl. Gel filtration buffer contained 150 mM NaCl.
Samples were injected onto either a Superose 6 or a Superdex 200 column (GE Healthcare) connected to an AKTA liquid chromatography system. Fractions of 0.5 ml were collected and re-suspended in Laemmli sample buffer, fractionated by SDS-PAGE and analyzed by western blotting. Endogenous Trs33p, Bet3p and Trs23p were detected using polyclonal antibodies raised in rabbits, the HA epitope was detected using a commercially available monoclonal antibody (HA.7, Sigma), the myc epitope was detected using the monoclonal antibody 9E10 and the TAP tag was detected with anti-TAP antibody (OpenBiosystems).
Recombinant protein
Yeast TRAPP subunits were expressed and purified from E. coli BL21(DE3) cells as previously described.Citation15 Bet3p and Trs33p subunits were 6xHis-tagged and complexes were purified using Ni2+-NTA (Qiagen) followed by size-exclusion chromatography on a Superdex 200 column.
Bacterial lysates
Yeast TRAPP subunits were expressed in E. coli BL21 (DE3) cells as for recombinant protein (see above). Cells were sonicated in lysis buffer (20 mM HEPES pH 7.3, 1 mM DTT) and the lysates were clarified by centrifugation at 30,000 g in a JA25.50 rotor (Beckman) prior to their use in guanine nucleotide exchange factor assays.
Guanine nucleotide exchange factor (GEF) assays
GEF assays were performed as previously described.Citation19 Each reaction was started in a volume of 80 μl with yeast or bacterial lysates added to a final concentration of 4 mg/ml. For Ypt1p alone, bovine serum albumin (BSA) or E. coli BL21(DE3) lysate was added to a final concentration of 4 mg/ml. The zero minute time point for each sample was taken immediately after the pre-loaded Rab was added and 18 μl was removed for each subsequent time point. Percent 3H-GDP remaining was based on the individual zero for each sample.
In vivo and in vitro trafficking assays
The CPY pulse chase experiment was performed as previously described with some modifications.Citation58 Five OD600 units of cells in early log phase (OD600 1–1.5) were temperature shifted in a shaking 38.5°C water bath for 30 min. Cells were labeled with 250 μCi of 35S-methionine/cysteine (Promix, Perkin-Elmer) (pulsed) for 4 min followed by the addition of cold methionine/cysteine (chased) at a final concentration of 10 mM. The zero time point was taken immediately after the cold methionine/cysteine was added. Cells were chased for a total of 30 min with an equal volume of cells being removed every 10 min. Cells from each time point were converted to spheroplasts, lysed and CPY was immunoprecipitated overnight using a rabbit polyclonal anti-CPY antibody (Abcam). Immunoprecipitated CPY was fractionated by SDS-PAGE and the amounts loaded were normalized to the counts per minute measured from whole cell lysates. The gel was Coomassie-stained, dried and labeled CPY was detected by autoradiography.
Microscopy-based assays (GFP-Snc1p, GFP-Ypt31p) were performed by transforming yeast with the appropriate plasmid. Cells were grown to early log phase overnight at 25°C and 5 OD600 units of cells were resuspended in 1 ml of 3.7% paraformaldehyde (PFA), incubated at room temperature for 15 min, pelleted and resuspended in ice cold methanol for 5 min. Cells were then washed with wash buffer (1.4 M sorbitol, 50 mM KPi, pH 7.5) and resuspended in 100 μl of the same buffer. Cells were visualized using a Zeiss axioplan microscope fitted with an X-cite series 120Q light source (EXFO Life Sciences) and a Lumenera Infinity 3–1C 1.4 megapixel cooled CCD camera.
The in vitro reconstituted ER-to-Golgi assay using purified proteins was performed as described.Citation34
Maturation of Ape1p-GFP was performed by first transforming the plasmid into yeast. The cells were then grown overnight to early log phase in minimal medium lacking uracil at which point they were converted to spheroplasts and lysed in 20 mM HEPES, pH 7.3, 100 mM NaCl, 0.1 mM DTT, 2 mM EDTA with protease inhibitors. Equal amounts of protein were fractionated by SDS-PAGE, transferred to PVDF membrane and probed with mouse anti-GFP (Roche). Quantitation was performed using Image J freeware.
Yeast two-hybrid assay
TRS23 and variants of this protein were cloned into a Gateway®-modified pGBKT7 vector (pGBKT7GTWY).Citation21 All other TRAPP subunits were cloned into the pGADT7 vector by restriction enzyme cloning. Mating and analysis were as described by Scrivens et al.Citation21
Chemical crosslinking
Fractionated samples in gel filtration buffer (50 mM NaCl) were treated with 2 mM dithiobis[succinimidyl] propionate (DSP) for 3 h on ice. The reaction was quenched by adding Tris pH 7.5 to a final concentration of 20 mM and incubating at room temperature for 15 min. For immunoprecipitation, the sample was diluted with gel filtration buffer (100 mM NaCl) containing 1% Triton X-100 and incubated with 2 μg IgG overnight on ice. The next day, immune complexes were collected on protein A beads, washed with lysis buffer and processed for western analysis. In some experiments the crosslinked samples were re-fractionated on a Superose 6 column as described above.
Acknowledgments
We are grateful to Dr Ruth Collins for the GFP-Ypt31p construct, Dr Hugh Pelham for the GFP-Snc1p construct, Dr Randy Schekman for the sec18 mutant, Dr Dan Klionsky for the Ape1p-GFP construct and Dr Vladimir Titorenko for providing the P. pastoris strain. We thank members of the Sacher laboratory and Drs Christopher Brett and Malcolm Whiteway for helpful comments, and Mr Ming Yang Sun for help in the preparation of . The initial random mutagenesis of TRS23 was performed at the 2007 Cold Spring Harbor Laboratory Yeast Genetics course. During the course of this study, S.B. was supported by a Canadian Institutes of Health Research (CIHR) award and is presently a recipient of a Fonds Que´be´cois de la Recherche sur la Nature et les Technologies doctoral award. M.S. is a recipient of a CIHR New Investigator award. This study was supported by CIHR, the Natural Sciences and Engineering Research Council and the Canada Foundation for Innovation to M.S. M.S. is a member of the Groupe de Recherche Axe´ sur la Structure des Prote´ines (GRASP).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Lee MC, Miller EA. Molecular mechanisms of COPII vesicle formation. Semin Cell Dev Biol 2007; 18:424 - 34; http://dx.doi.org/10.1016/j.semcdb.2007.06.007; PMID: 17686639
- Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell 2007; 12:671 - 82; http://dx.doi.org/10.1016/j.devcel.2007.04.005; PMID: 17488620
- Sztul E, Lupashin V. Role of vesicle tethering factors in the ER-Golgi membrane traffic. FEBS Lett 2009; 583:3770 - 83; http://dx.doi.org/10.1016/j.febslet.2009.10.083; PMID: 19887069
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993; 362:318 - 24; http://dx.doi.org/10.1038/362318a0; PMID: 8455717
- Gillingham AK, Munro S. Long coiled-coil proteins and membrane traffic. Biochim Biophys Acta 2003; 1641:71 - 85; http://dx.doi.org/10.1016/S0167-4889(03)00088-0; PMID: 12914949
- Yu IM, Hughson FM. Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol 2010; 26:137 - 56; http://dx.doi.org/10.1146/annurev.cellbio.042308.113327; PMID: 19575650
- Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, et al. TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Mol Cell 2001; 7:433 - 42; http://dx.doi.org/10.1016/S1097-2765(01)00190-3; PMID: 11239471
- Sacher M, Kim YG, Lavie A, Oh BH, Segev N. The TRAPP complex: insights into its architecture and function. Traffic 2008; 9:2032 - 42; http://dx.doi.org/10.1111/j.1600-0854.2008.00833.x; PMID: 18801063
- Cai H, Zhang Y, Pypaert M, Walker L, Ferro-Novick S. Mutants in trs120 disrupt traffic from the early endosome to the late Golgi. J Cell Biol 2005; 171:823 - 33; http://dx.doi.org/10.1083/jcb.200505145; PMID: 16314430
- Lynch-Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, et al. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci U S A 2010; 107:7811 - 6; http://dx.doi.org/10.1073/pnas.1000063107; PMID: 20375281
- Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, et al. TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J 1998; 17:2494 - 503; http://dx.doi.org/10.1093/emboj/17.9.2494; PMID: 9564032
- Meiling-Wesse K, Epple UD, Krick R, Barth H, Appelles A, Voss C, et al. Trs85 (Gsg1), a component of the TRAPP complexes, is required for the organization of the preautophagosomal structure during selective autophagy via the Cvt pathway. J Biol Chem 2005; 280:33669 - 78; http://dx.doi.org/10.1074/jbc.M501701200; PMID: 16079147
- Nazarko TY, Huang J, Nicaud JM, Klionsky DJ, Sibirny AA. Trs85 is required for macroautophagy, pexophagy and cytoplasm to vacuole targeting in Yarrowia lipolytica and Saccharomyces cerevisiae. Autophagy 2005; 1:37 - 45; http://dx.doi.org/10.4161/auto.1.1.1512; PMID: 16874038
- Cai Y, Chin HF, Lazarova D, Menon S, Fu C, Cai H, et al. The structural basis for activation of the Rab Ypt1p by the TRAPP membrane-tethering complexes. Cell 2008; 133:1202 - 13; http://dx.doi.org/10.1016/j.cell.2008.04.049; PMID: 18585354
- Kim YG, Raunser S, Munger C, Wagner J, Song YL, Cygler M, et al. The architecture of the multisubunit TRAPP I complex suggests a model for vesicle tethering. Cell 2006; 127:817 - 30; http://dx.doi.org/10.1016/j.cell.2006.09.029; PMID: 17110339
- Sacher M, Barrowman J, Schieltz D, Yates JR 3rd, Ferro-Novick S. Identification and characterization of five new subunits of TRAPP. Eur J Cell Biol 2000; 79:71 - 80; http://dx.doi.org/10.1078/S0171-9335(04)70009-6; PMID: 10727015
- Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, et al. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature 2007; 445:941 - 4; http://dx.doi.org/10.1038/nature05527; PMID: 17287728
- Jones S, Newman C, Liu F, Segev N. The TRAPP complex is a nucleotide exchanger for Ypt1 and Ypt31/32. Mol Biol Cell 2000; 11:4403 - 11; PMID: 11102533
- Wang W, Sacher M, Ferro-Novick S. TRAPP stimulates guanine nucleotide exchange on Ypt1p. J Cell Biol 2000; 151:289 - 96; http://dx.doi.org/10.1083/jcb.151.2.289; PMID: 11038176
- Choi C, Davey M, Schluter C, Pandher P, Fang Y, Foster LJ, et al. Organization and assembly of the TRAPPII complex. Traffic 2011; 12:715 - 25; http://dx.doi.org/10.1111/j.1600-0854.2011.01181.x; PMID: 21453443
- Scrivens PJ, Noueihed B, Shahrzad N, Hul S, Brunet S, Sacher M. C4orf41 and TTC-15 are mammalian TRAPP components with a role at an early stage in ER-to-Golgi trafficking. Mol Biol Cell 2011; 22:2083 - 93; http://dx.doi.org/10.1091/mbc.E10-11-0873; PMID: 21525244
- Yamasaki A, Menon S, Yu S, Barrowman J, Meerloo T, Oorschot V, et al. mTrs130 is a component of a mammalian TRAPPII complex, a Rab1 GEF that binds to COPI-coated vesicles. Mol Biol Cell 2009; 20:4205 - 15; http://dx.doi.org/10.1091/mbc.E09-05-0387; PMID: 19656848
- Fan S, Feng Y, Wei Z, Xia B, Gong W. Solution structure of synbindin atypical PDZ domain and interaction with syndecan-2. Protein Pept Lett 2009; 16:189 - 95; http://dx.doi.org/10.2174/092986609787316342; PMID: 19200043
- Fan S, Wei Z, Xu H, Gong W. Crystal structure of human synbindin reveals two conformations of longin domain. Biochem Biophys Res Commun 2009; 378:338 - 43; http://dx.doi.org/10.1016/j.bbrc.2008.04.143; PMID: 18466758
- Yip CK, Berscheminski J, Walz T. Molecular architecture of the TRAPPII complex and implications for vesicle tethering. Nat Struct Mol Biol 2010; 17:1298 - 304; http://dx.doi.org/10.1038/nsmb.1914; PMID: 20972447
- Menon S, Cai H, Lu H, Dong G, Cai Y, Reinisch K, et al. mBET3 is required for the organization of the TRAPP complexes. Biochem Biophys Res Commun 2006; 350:669 - 77; http://dx.doi.org/10.1016/j.bbrc.2006.09.096; PMID: 17027922
- Montpetit B, Conibear E. Identification of the novel TRAPP associated protein Tca17. Traffic 2009; 10:713 - 23; http://dx.doi.org/10.1111/j.1600-0854.2009.00895.x; PMID: 19220810
- Morozova N, Liang Y, Tokarev AA, Chen SH, Cox R, Andrejic J, et al. TRAPPII subunits are required for the specificity switch of a Ypt-Rab GEF. Nat Cell Biol 2006; 8:1263 - 9; http://dx.doi.org/10.1038/ncb1489; PMID: 17041589
- Esters H, Alexandrov K, Iakovenko A, Ivanova T, Thomä N, Rybin V, et al. Vps9, Rabex-5 and DSS4: proteins with weak but distinct nucleotide-exchange activities for Rab proteins. J Mol Biol 2001; 310:141 - 56; http://dx.doi.org/10.1006/jmbi.2001.4735; PMID: 11419942
- Jiang Y, Scarpa A, Zhang L, Stone S, Feliciano E, Ferro-Novick S. A high copy suppressor screen reveals genetic interactions between BET3 and a new gene. Evidence for a novel complex in ER-to-Golgi transport. Genetics 1998; 149:833 - 41; PMID: 9611195
- Lewis MJ, Nichols BJ, Prescianotto-Baschong C, Riezman H, Pelham HR. Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol Biol Cell 2000; 11:23 - 38; PMID: 10637288
- Buvelot Frei S, Rahl PB, Nussbaum M, Briggs BJ, Calero M, Janeczko S, et al. Bioinformatic and comparative localization of Rab proteins reveals functional insights into the uncharacterized GTPases Ypt10p and Ypt11p. Mol Cell Biol 2006; 26:7299 - 317; http://dx.doi.org/10.1128/MCB.02405-05; PMID: 16980630
- Shintani T, Huang WP, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell 2002; 3:825 - 37; http://dx.doi.org/10.1016/S1534-5807(02)00373-8; PMID: 12479808
- Barlowe C. Coupled ER to Golgi transport reconstituted with purified cytosolic proteins. J Cell Biol 1997; 139:1097 - 108; http://dx.doi.org/10.1083/jcb.139.5.1097; PMID: 9382859
- Sacher M, Ferro-Novick S. Purification of TRAPP from Saccharomyces cerevisiae and identification of its mammalian counterpart. Methods Enzymol 2001; 329:234 - 41; http://dx.doi.org/10.1016/S0076-6879(01)29083-1; PMID: 11210539
- Scrivens PJ, Shahrzad N, Moores A, Morin A, Brunet S, Sacher M. TRAPPC2L is a novel, highly conserved TRAPP-interacting protein. Traffic 2009; 10:724 - 36; http://dx.doi.org/10.1111/j.1600-0854.2009.00906.x; PMID: 19416478
- Loh E, Peter F, Subramaniam VN, Hong W. Mammalian Bet3 functions as a cytosolic factor participating in transport from the ER to the Golgi apparatus. J Cell Sci 2005; 118:1209 - 22; http://dx.doi.org/10.1242/jcs.01723; PMID: 15728249
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12:831 - 5; http://dx.doi.org/10.1038/ncb0910-831; PMID: 20811355
- Schweizer A, Fransen JA, Matter K, Kreis TE, Ginsel L, Hauri HP. Identification of an intermediate compartment involved in protein transport from endoplasmic reticulum to Golgi apparatus. Eur J Cell Biol 1990; 53:185 - 96; PMID: 1964413
- Schmidt K, Stephens DJ. Cargo loading at the ER. Mol Membr Biol 2010; 27:398 - 411; http://dx.doi.org/10.3109/09687688.2010.506203; PMID: 21142873
- Yu S, Satoh A, Pypaert M, Mullen K, Hay JC, Ferro-Novick S. mBet3p is required for homotypic COPII vesicle tethering in mammalian cells. J Cell Biol 2006; 174:359 - 68; http://dx.doi.org/10.1083/jcb.200603044; PMID: 16880271
- Bacon RA, Salminen A, Ruohola H, Novick P, Ferro-Novick S. The GTP-binding protein Ypt1 is required for transport in vitro: the Golgi apparatus is defective in ypt1 mutants. J Cell Biol 1989; 109:1015 - 22; http://dx.doi.org/10.1083/jcb.109.3.1015; PMID: 2504726
- Jedd G, Richardson C, Litt R, Segev N. The Ypt1 GTPase is essential for the first two steps of the yeast secretory pathway. J Cell Biol 1995; 131:583 - 90; http://dx.doi.org/10.1083/jcb.131.3.583; PMID: 7593181
- Sclafani A, Chen S, Rivera-Molina F, Reinisch K, Novick P, Ferro-Novick S. Establishing a role for the GTPase Ypt1p at the late Golgi. Traffic 2010; 11:520 - 32; http://dx.doi.org/10.1111/j.1600-0854.2010.01031.x; PMID: 20059749
- McNew JA, Sogaard M, Lampen NM, Machida S, Ye RR, Lacomis L, et al. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J Biol Chem 1997; 272:17776 - 83; http://dx.doi.org/10.1074/jbc.272.28.17776; PMID: 9211930
- Tsui MM, Tai WC, Banfield DK. Selective formation of Sed5p-containing SNARE complexes is mediated by combinatorial binding interactions. Mol Biol Cell 2001; 12:521 - 38; PMID: 11251068
- Ungermann C, von Mollard GF, Jensen ON, Margolis N, Stevens TH, Wickner W. Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J Cell Biol 1999; 145:1435 - 42; http://dx.doi.org/10.1083/jcb.145.7.1435; PMID: 10385523
- von Mollard GF, Nothwehr SF, Stevens TH. The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. J Cell Biol 1997; 137:1511 - 24; http://dx.doi.org/10.1083/jcb.137.7.1511; PMID: 9199167
- Boyd C, Hughes T, Pypaert M, Novick P. Vesicles carry most exocyst subunits to exocytic sites marked by the remaining two subunits, Sec3p and Exo70p. J Cell Biol 2004; 167:889 - 901; http://dx.doi.org/10.1083/jcb.200408124; PMID: 15583031
- Pokrovskaya ID, Willett R, Smith RD, Morelle W, Kudlyk T, Lupashin VV. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011; 21:1554 - 69; http://dx.doi.org/10.1093/glycob/cwr028; PMID: 21421995
- Vasile E, Oka T, Ericsson M, Nakamura N, Krieger M. IntraGolgi distribution of the Conserved Oligomeric Golgi (COG) complex. Exp Cell Res 2006; 312:3132 - 41; http://dx.doi.org/10.1016/j.yexcr.2006.06.005; PMID: 16857184
- Stagg SM, Gürkan C, Fowler DM, LaPointe P, Foss TR, Potter CS, et al. Structure of the Sec13/31 COPII coat cage. Nature 2006; 439:234 - 8; http://dx.doi.org/10.1038/nature04339; PMID: 16407955
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, et al. Molecular anatomy of a trafficking organelle. Cell 2006; 127:831 - 46; http://dx.doi.org/10.1016/j.cell.2006.10.030; PMID: 17110340
- Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, et al. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature 2011; 473:181 - 6; http://dx.doi.org/10.1038/nature09969; PMID: 21532587
- Conibear E. Vesicle transport: springing the TRAPP. Curr Biol 2011; 21:R506 - 8; http://dx.doi.org/10.1016/j.cub.2011.05.045; PMID: 21741587
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998; 14:953 - 61; http://dx.doi.org/10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U; PMID: 9717241
- Kim DW, Sacher M, Scarpa A, Quinn AM, Ferro-Novick S. High-copy suppressor analysis reveals a physical interaction between Sec34p and Sec35p, a protein implicated in vesicle docking. Mol Biol Cell 1999; 10:3317 - 29; PMID: 10512869
- Rossi G, Kolstad K, Stone S, Palluault F, Ferro-Novick S. BET3 encodes a novel hydrophilic protein that acts in conjunction with yeast SNAREs. Mol Biol Cell 1995; 6:1769 - 80; PMID: 8590804