Abstract
There are numerous experimental approaches to identify the interaction networks of soluble proteins, but strategies for the identification of membrane protein interactomes remain limited. We discuss in detail the logic of an experimental design that led us to identify the interactome of a membrane protein of complex membrane topology, the calcium activated chloride channel Anoctamin 1/Tmem16a (Ano1). We used covalent chemical stabilizers of protein-protein interactions combined with magnetic bead immuno-affinity chromatography, quantitative SILAC mass-spectrometry and in silico network construction. This strategy led us to define a putative Ano1 interactome from which we selected key components for functional testing. We propose a combination of procedures to narrow down candidate proteins interacting with a membrane protein of interest for further functional studies.
Biological membranes are interfaces between environments of differing composition. This membrane function is dynamic as membranes are conduits that interpret and modify their surroundings. Nowhere is this clearer than in the plasma membrane where fundamental cell processes critical for preserving cellular homeostasis take place. In both unicellular and multicellular organisms, plasma membrane proteins play integral roles in cell signaling, cell-cell and cell-matrix adhesion, transport of molecules and ions, cellular metabolism and maintenance of cellular architecture. All of these cellular functions critically depend on the unique lipid and protein composition of membranes. Here we focus on the problem of how to determine protein-protein associations that membrane proteins establish in their transmembrane, exofacial and endofacial domains in order to perform their functions. We use as a case study the calcium activated chloride channel Anoctamin 1 (Ano1).Citation1
Genomic studies suggest that membrane proteins make up one quarter or more of all the predicted proteins in an organism. This fraction seems conserved from prokaryotes to Homo sapiens.Citation2-Citation4 Apart from their abundance, the extensive and diverse roles that membrane proteins play in cell-cell adhesion, intracellular and intercellular transport, cell-cell recognition and anchoring to cytoskeleton showcase their significance. Mutations in membrane proteins are implicated in a wide number of pathologies including heart disease, kidney dysfunction, myotonias, tumorigenesis, cystic fibrosis and an array of neuropathologies.Citation5
Nearly 60% of all FDA approved drugs target membrane proteins. In contrast, therapeutic drugs that target nuclear or cytoplasmic proteins account each for a mere 6%.Citation6,Citation7 Prominently represented in “druggable” membrane proteins are G-protein-coupled receptors and ion channels.Citation6,Citation7 G-protein-coupled receptors are the largest family of drug-susceptible targets.Citation7 The numbers of G-protein-coupled receptors are estimated to be ~700–800 out of ~2,600 genes in the human genome (Homo sapiens build 37.3 consensus coding sequence (CCDS) July 2012Citation8).Citation9-Citation11 The third most abundant is the ion channel family of proteins with at least 232 genes that encode pore-forming plasma membrane ion channels.Citation11-Citation13 Most of these “druggable” membrane proteins adopt complex membrane topologies making their purification and the identification of their interactors challenging. Ano1, the focus of this addendum, belongs to the channel family and its activity is susceptible to chemical inhibitors. Moreover, Ano1 possesses a complex membrane insertion topology making it a good candidate to test new approaches for membrane protein biochemistry scalable to other membrane proteins ().
Figure 1. Membrane topology of Ano1. Model of mouse Ano1 topology. This model is based on data from Yu et al. (2012).Citation14 In this model the sequence 628–638 forms the outer vestibule of the channel and amino acids in the first and third intracellular loops are involved in Ca-dependent gating. See Yu et al. for further details.Citation14

The Paucity of Membrane Protein Interaction Networks
Although membrane proteins such as G-protein-coupled receptors and ion channels are abundant and critical, our knowledge of how these proteins are organized in the membrane with other proteins and how they couple to extracellular and intracellular proteins is far from comprehensive. Although great progress has been made in recent years in purification of membrane proteins for crystallography, these approaches involve disruption of the native membrane environment and replacement with artificial lipids. To understand how a membrane protein co-exists with other proteins, one of the main hurdles is to reproduce a milieu that preserves the protein's inherent function, structure and protein-protein interactions. These difficulties have led to a major underrepresentation of membrane proteins in protein-interaction networks or interactomes. Here we use a wide definition of interactome as the network of molecular, genetic and/or metabolic associations aiming at describing contents, structure, function, behavior, or combinations thereof either of a protein, pathway, organelle, or cell.Citation15
Most protein-protein interactions that have been discovered for G-protein-coupled receptors and channels have been defined painstakingly one-by-one or few at a time. Comprehensive genome- or proteome-wide strategies have been used relatively little for membrane proteins. Of nearly 700 human G-protein-coupled receptors that have been curated, fewer than 100 interacting proteins have been identified for less than 10% of them.Citation16 The picture is even more fragmented for vertebrate ion channels except for few exceptions.Citation17 In contrast, large-scale screening for pairwise interactions among 705 Saccharomyces cerevisiae proteins annotated as integral membrane proteins has provided nearly 500 protein interactors that participate in ~2,000 putative protein-protein interactions.Citation18 This simple comparison tells us that protein interactions networks of vertebrate membrane proteins are under-explored. Except for Saccharomyces cerevisiae, this is a global problem that spans phyla. Analysis of curated protein-protein interactions gathered in the BIOGRID database of protein-protein interactions indicates that less than one-third of all reported interactions include membrane proteins.Citation19 Moreover there is a significant under-representation of GO categories linked to the membrane: “intrinsic to membrane,” “integral to membrane,” “Golgi apparatus” and “membrane part.”Citation19 Under-representation is irrespective of the method used to identify protein-protein interactions such as affinity capture followed by mass-spectrometry or immunoblot or yeast-two hybrid system.Citation19 These systemic problems with membrane proteins have been approached successfully in several ways. For example, yeast-two hybrid strategies have been used where the bait and prey association are either excluded from the nucleus (protein fragment complementation)Citation20 or constrained to a membrane such as in the split-Ubiquitin membrane yeast-two hybridCitation18 or more recently by tandem affinity purification of tagged membrane proteins from yeast detergent extracts. Alternatively, identifying interactions at the gene level provide a proxy for protein-protein associations since one-fifth to one-tenth of protein-protein interaction pairs have a correlate in a genetic interaction pair.Citation21
Philosophy of the Approach Presented Here
There are diverse challenges facing the investigator in pursuit of a membrane protein interactome. Among them are: (1) the low relative abundance of membrane proteins, (2) the need for membrane solubilization with detergents, which may only partially solubilize membrane proteins or alter membrane protein function and their interactionsCitation22 and (3) the challenge of selecting candidates for study after protein identification by mass spectrometry, which routinely provides hundreds of proteins for one experiment. The approach presented here intends to minimize some of these problems and provide a rational approach, based on quantitative proteomics and in silico tools, to select candidates for study among the hundreds proteins detected by sensitive mass spectrometry methods. Even with these goals in mind, researchers should be aware that solving one problem might create others. For example, to overcome the low abundance of the membrane protein that we focused on, Ano1, we resorted to recombinant expression. This opens the door for new problems such as potential mislocalization due to overexpression and biases in defining protein associated stoichiometries.
As with the interpretation of any large data set, the approach presented here provides a prioritized list of potential interactors. Claims of “physiological relevance” ought to be founded in multipronged confirmation of each individual interaction. Moreover, it is central to emphasize that there is no substitution for confirmatory functional studies of a putative protein-protein interaction pair using endogenously expressed proteins in a tissue of interest.
Identification of a Membrane Protein Interactome: the Ano1 Case Study with its Advantages and Caveats
We chose to focus on the calcium activated chloride channel Ano1Citation23-Citation27 (). Ano1 is expressed in the apical domain of exocrine cellsCitation28,Citation29 like salivary glands and is essential for salivary exocrine secretion () and a growing list of other functions.Citation31 Members of the anoctamin family play central physiological roles in a multitude of cells including but not limited to epithelial secretion,Citation32 signal transduction in sensory systems,Citation33-Citation37 modulation of smooth muscle contractionCitation38 and control of neuronal and cardiac excitability.Citation39 Ano1 is an eight transmembrane domain channel (). Ano1 molecular interactions through its intramembrane, exofacial and endofacial domains are mostly unknown.Citation26,Citation27 The complex membrane topology of Ano1, with multiple loops connecting Ano1’s trans-membrane domains, makes the identification of its interactome challenging as each hydrophilic segment of the protein would need to be tested individually in solution to identify its associated proteins (). Furthermore, Ano1, like other membrane proteins, may associate with accessory subunits that are themselves membrane proteins and interact within the plane of the lipid bilayer.
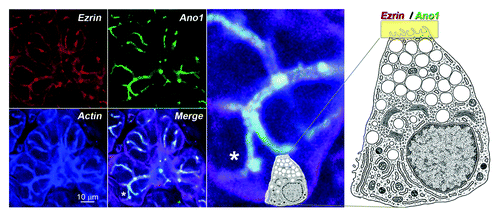
Figure 2. Subcellular localization of the Ano1 signaling complex. This complex is shown here by the colocalization of Ano1 (green), the acting binding signaling protein ezrin (red) and actin (blue) in the apical membranes of acinar and duct cells of the submaxillary gland of mouse. The white strings represent the canalicullar lumen highlighted by the overlapping fluorescent signals. The landmark asterisk in the merged panel is enlarged to the right. Cell diagram depicts a submaxillary secreting cell and its relative position in the acinus. A yellow band on the apical domain marks the localization of ezrin and Ano1. Diagram was modified from Lentz.Citation30 Bar 10 microns.
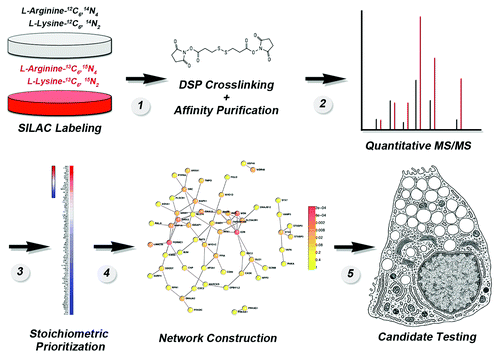
We assembled a sequence of well-established approaches to identify the putative interactome of Ano1. This approach could potentially be used to identify putative interactome of other membrane protein. briefly presents this experimental sequence. Two cell lines are used. One expresses a functional tagged membrane protein while the control is identical except that it lacks the recombinant membrane protein. One of these cell lines is labeled with either regular mediun (, gray plate) while the second is labeled with non-radioactive isotope-labeled lysine and arginine-containing medium (, red plate) to equilibrium (this typically requires growth in labeling medium for six generations or more). Intact cells from each condition are then exposed to a membrane-permeant cross-linker, such as [dithiobis(succinimidyl propionate)] DSP, whose concentration is controlled to stabilize only close-proximity interactions (, step 1). The utility of using DSP is illustrated by the observation that the proteins co-purifying with Ano1 in the presence of DSP are reduced 15-fold when DSP is omitted.Citation1 However, the use of DSP or any other cross-linker has to be carefully weighted since their use may introduce new variables that could confound data. For example, cross-linkers used at high concentrations may lead to simply fixing the whole cell.Citation40 In contrast, cross-linkers at low concentration can be inefficient leading to low percentage of stabilized protein complexes and an apparently low stoichiometry of interaction. To overcome these limitations, we defined concentrations, time and temperature of cross-linker incubation in vivo so that cross-linking increases by a 2- to 3-fold the bait’s molecular size, as determined by sucrose sedimentation. This provides a biochemical indication that the cross-linker has not created extended networks of protein interactions. For example, Ano1 sediments at ~6S without cross-linking yet after DSP treatment the shift in the sedimentation represents an increase of at least ~3S.Citation1 This increase in the sedimentation coefficient would roughly correspond to a doubling of the molecular size of a ~150 kDa protein such as Ano1. This has been a universal criterion used in all our work with cross-linkers to minimize the possibility of false-positives due to extended networks of cross-linked proteins.Citation1,Citation41,Citation42
Figure 3. Flow diagram of the strategy to generate an in vivo membrane protein interactome using SILAC mass spectrometry. See text for details and Perez-Cornejo et al. (2012).Citation1
Detergent-soluble cell extracts are applied to immunoaffinity magnetic bead matrices, where they are retained due to the inclusion of antibodies directed against the tag on the recombinant protein. The bound complexes are eluted with a molar excess of a peptide corresponding to the tag (, step 1). Eluates from matrices incubated with transfected cell (“heavy” isotope labeled) and non-transfected cell (unlabeled “light”) extracts are mixed 1:1 and analyzed by MS/MS (, step 2). Mass spectrometry determines peptide identity, distinguishes peptides based on their isotope label and provides an enrichment of peptides between the two experimental conditions, which can be differentiated by the isotope tags in them. Putative interacting proteins are prioritized based on their fold enrichment as a proxy for stoichiometry of association (, steps 2–3). Prioritized proteins are organized into putative networks (, step 4), and the network nodes with the highest number of connections are selected for functional testing (, step 5). We asked that this experimental strategy satisfy the traditional biochemical criteria of minimizing false positive protein-protein interactions common to mass-spectrometry approaches.Citation43,Citation44 For example, we identified the proteome that binds to antibody-bead matrices and we subtracted it from the Ano1 proteome. The outcome of this subtraction is quite illustrative since the Ano1 proteome led to the identification of 509 proteins, which are reduced to 209 proteins after subtracting the non-specific bead proteome. Of these 209 proteins only 93 are enriched more than 2-fold by SILAC enrichment.Citation1 In addition, our approach also considers the following:
(1) Protein isolation and identification should be quantitative.
(2) Protein isolation and identification should consider the contribution of lipid bilayers to the native state and interactions of a membrane protein.
(3) Experimental strategy should be designed with the realization that the bait membrane protein may reside in different membrane compartments either because of protein trafficking or overexpression artifacts.
(4) The strategy should be adaptable for the identification of sub-proteomes.
Protein isolation and identification should take advantage of quantitative approaches to binding partner identification and sorting
This criterion is a critical step in selecting and narrowing down candidates after a high-throughput screen has delivered a large number of “hits” (). This step is central for subsequently designing experiments to test the functionality of individual protein-protein interactions. We prefer SILAC, (stable isotope labeling with amino acids in culture) to quantitatively label the whole proteome of a cellCitation45-Citation48 followed by immuno-affinity purification (using the membrane protein of interest as the bait protein) with magnetic beads and the use of antigenic peptides for elutionCitation1,Citation41,Citation42,Citation49 (). This method offers three major advantages. First, isotope labeling occurs in vivo and the whole proteome is uniformly tagged while the native state of proteins and their interactions is maintained. Second, the proteomes can be distinguished by the isotopic tags in lysine and arginine allowing a contrast between two experimental conditions (). In our Ano1 experiments, we used this to define those proteins that spuriously bind to magnetic bead immuno-affinity matrices, but the same approach could be used to compare the interactomes of different splice variants, for example. Finally, the fold enrichment prioritizes components in the putative interactome (). This is of great advantage as we can estimate apparent stoichiometries among proteins in the interactome when compared with the bait. We qualify the stoichiometry as “apparent” because the bait, Ano1, is recombinantly expressed likely above the levels of the endogenous protein, a fact that necessarily biases stoichiometries. For example the recombinant Ano1 interactome identified 93 proteins enriched greater than 2-fold.Citation1 Ano1 itself was enriched ~30 fold and 4 of the identified proteins were similarly enriched ~30 fold. These top proteins co-enriching with Ano1 are the actin-binding proteins Ezrin, Moesin, Radixin,Citation50,Citation51 which we functionally confirmed as modulators of Ano1 channel activity.Citation1 In contrast, a protein in the interactome data set that is found enriched only 2-fold may be lower due to its expression in cells at levels below the recombinant target (Ano1-FLAG), localization to distinct cellular places at steady-state, lower affinity, lack of optimal cross-linking, or other reasons, but may still represent a potential biologically relevant interactor.
Stoichiometrically prioritized interactomes can still be composed of dozens or hundreds of potential interactors. This makes the selection of candidate proteins for experimentation problematic. Thus, we coupled the Ano1 protein interactome to an in silico data mining algorithm to generate a proposed protein interaction network (). The proposed interaction network built in silico is then used to select high connectivity proteins for biochemical and functional testing. It is important to highlight that this putative network does not represent the actual map of experimentally verified functional protein-protein interactions. Rather, it takes advantage of an independently generated data set that can be used to refine the interactome data set to select those candidates most likely to represent physiologically relevant interactors. To construct a network, we preferred the Dapple algorithm due its predictive value, its statistical definition and ranking of network nodes and architecture and the stringency of the protein-protein interaction databases selected (www.broadinstitute.org/mpg/dapple/dapple.php).Citation52 DAPPLE builds protein interactions networks gathering significant physical connectivity among proteins from protein-protein interactions databases (MINT, BIND, IntAct, PPrel, ECrel, Reactome and others). The data set contains nearly half a million interactions across ~13,000 proteins.Citation52 The fundamental premise of using this algorithm is the observation that the magnitude and severity of phenotypes observed in null alleles affecting the gene of any given protein are more pronounced among proteins with the highest number of protein-protein connections in a network.Citation53 Thus, we inferred that the magnitude of possible phenotypes generated by elimination of a protein in the putative Ano1 network should be most pronounced among the most connected proteins. This was the case of Ezrin, Moesin, Radixin in the Ano1 interactome where these proteins establish five connections with other proteins identified in the interactome.Citation1 This value is well above the average ~2 protein-protein connections observed in genome-wide interactomesCitation54 and the average 2.7 protein-protein connections of the Ano1 interactome.
Protein isolation and identification should consider the contribution of lipid bilayers to the native state and interactions of a membrane protein
The lipid environment is a key determinant in interactions between a membrane protein and cytosolic factors. This is exemplified by the lipid dependence of the association between sorting signals on membrane proteins and clathrin adaptors that recognize those sorting signals. Membrane phosphatidylinositol phospholipids increase the affinity of a clathrin adaptor AP-2 for a tyrosine sorting signals by ~2–3 orders of magnitude.Citation55,Citation56 This clearly illustrates the influence of the bilayer context in strengthening otherwise low affinity protein-protein interactions. Disruption of the membrane during solubilization of membrane proteins for analysis by immunoprecipitation is likely to result in the loss of these low-affinity, but potentially important, protein-protein interactions. To circumvent this problem, we used controlled protein crosslinking in intact cells with the cell-permeant crosslinker DSP [dithiobis(succinimidyl propionate)] (). The stabilization resulting from covalent crosslinking persists even after the necessary step of membrane dissolution with detergents, required for protein purification. DSP has a 12 Å spacer arm and a reversible disulfide bond that is cleaved with reducing agents to release cross-linked proteins after co-IP.Citation42,Citation49,Citation57 We have shown that DSP is suitable for a labile membrane protein-clathrin adaptor interaction.Citation58 Moreover, we have validated the use of DSP by genetically testing putative interactors.Citation41,Citation58 Critical to the use of DSP is to control the extent of the chemical crosslinking reaction to ~10–20% of the total bait protein present in the sample. Alternatively, we limit the increase of the bait original molecular size to 2–3 fold after cross-linking.Citation1 This criterion decreases, but does not eliminate, the chance of false-positive protein identification due to extensively crosslinked complexes expanding beyond proteins in the near vicinity of the bait. DSP is one of few cross-linker options whose chemistry is amenable for in vivo controlled protein crosslinking. Other cross-linkers can be tested for any membrane protein of interest yet their concentrations, temperature and time of cross-linking must be determined empirically and cannot be assumed similar between cross-linkersCitation59 (www.piercenet.com/browse.cfm?fldID=0203). Finally, while reversible crosslinking of proteins in situ is a powerful method to stabilize transient, weak, or low abundance protein-protein interactions it is central to keep in mind that the final definition of a protein-protein interaction pair requires multipronged experimental scrutiny.
Experimental strategy should be designed with the realization that the bait membrane protein may reside in different membrane compartments either because of protein traffic or overexpression artifacts
At the steady-state, membrane proteins usually are concentrated in one subcellular compartment, but during their life cycle they usually traffic along the endomembrane system of eukaryotic cells. Proteins resident in the plasma membrane can be internalized and recycled back to the cell surface via carriers bound to the cell surface either from endosomes or the Golgi complex.Citation60,Citation61 Thus, protein interactions engaged by a membrane protein will likely be distinct in different compartments where they inherently reside or transit. Controlled in vivo cross-linking allows the identification of proteins irrespective of the compartment where they traffic or reside as suggested by the identification of VAMP-3, an endosomal SNARE that cycles between plasma membrane and endosomes,Citation62,Citation63 which is co-enriched nearly 20-fold with Ano1.Citation1 However, if the membrane protein is recombinantly expressed subcellular localization should not be inferred from bait interactors as they may reflect artifacts of overexpression. Subcellular, localization should be determined from experiments performed with endogenous proteins
The strategy should be adaptable for the identification of sub-proteomes
depicts an approach designed to identify the widest possible interactome for Ano1. However, the approach can be adapted for the identification of compartment-specific interactomes. Two examples illustrate this point. First, a tag placed in the exofacial domain of a membrane protein would allow antibody application to intact cells prior to cross-linker treatment. Thus, it would be possible to purify just the surface pool of membrane proteins or those membrane proteins endocytosed after specific times. Second, the use of different cross-linker chemistries could be used to separate interactomes. For example, using membrane-impermeant cross-linkers could differentiate the exofacial interactome. In general, sulfo-derivates of cross-linkers such as DSP are excellent options.
Conclusions
The arsenal of tools to explore soluble protein interactions and their functionality is vast, yet options for membrane proteins are limited. Here we discussed the logic of an approach used by us to identify the interactome of a membrane channel, Ano1. We combined the best available tools of immuno-affinity chromatography, quantitative mass-spectrometry, in silico analysis and the use of reversible covalent chemical stabilizers of protein-protein interaction to define a high confidence list of membrane protein interactions to be experimentally validated one-by-one by alternate means (). Although this is one of several approaches to identify membrane protein interactomes, its reproducibility, comprehensive nature, multiple built-in controls and the rapid turn-around time from deciding to do the experiment to obtaining prioritized putative interactors compel us to recommend this experimental design.
Acknowledgments
Supported by grants from the NIH GM60448 (H.C.H.), EY014852 (H.C.H.), NS42599 (V.F.), GM077569 (V.F.), Emory University Research Committee (H.C.H.), NEI training grant 5T32EY007092–25 (C.D.) and NIH FIRST program fellowship K12 GM000608 (A.G.). Additional support was provided by the Microscopy Core of the Emory Neuroscience NINDS Core Facilities Grant P30NS055077 and NEI Core Grant P30EY006360.
References
- Perez-Cornejo P, Gokhale A, Duran C, Cui Y, Xiao Q, Hartzell HC, et al. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc Natl Acad Sci U S A 2012; 109:10376 - 81; http://dx.doi.org/10.1073/pnas.1200174109; PMID: 22685202
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305:567 - 80; http://dx.doi.org/10.1006/jmbi.2000.4315; PMID: 11152613
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci 1998; 7:1029 - 38; http://dx.doi.org/10.1002/pro.5560070420; PMID: 9568909
- Xia Y, Lu LJ, Gerstein M. Integrated prediction of the helical membrane protein interactome in yeast. J Mol Biol 2006; 357:339 - 49; http://dx.doi.org/10.1016/j.jmb.2005.12.067; PMID: 16413578
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL. The human disease network. Proc Natl Acad Sci U S A 2007; 104:8685 - 90; http://dx.doi.org/10.1073/pnas.0701361104; PMID: 17502601
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there?. Nat Rev Drug Discov 2006; 5:993 - 6; http://dx.doi.org/10.1038/nrd2199; PMID: 17139284
- Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug-target network. Nat Biotechnol 2007; 25:1119 - 26; http://dx.doi.org/10.1038/nbt1338; PMID: 17921997
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 2009; 19:1316 - 23; http://dx.doi.org/10.1101/gr.080531.108; PMID: 19498102
- Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 2003; 63:1256 - 72; http://dx.doi.org/10.1124/mol.63.6.1256; PMID: 12761335
- Kroeze WK, Sheffler DJ, Roth BL. G-protein-coupled receptors at a glance. J Cell Sci 2003; 116:4867 - 9; http://dx.doi.org/10.1242/jcs.00902; PMID: 14625380
- Sharman JL, Mpamhanga CP, Spedding M, Germain P, Staels B, Dacquet C, et al, NC-IUPHAR. IUPHAR-DB: new receptors and tools for easy searching and visualization of pharmacological data. Nucleic Acids Res 2011; 39:Database issue D534 - 8; http://dx.doi.org/10.1093/nar/gkq1062; PMID: 21087994
- Jegla TJ, Zmasek CM, Batalov S, Nayak SK. Evolution of the human ion channel set. Comb Chem High Throughput Screen 2009; 12:2 - 23; http://dx.doi.org/10.2174/138620709787047957; PMID: 19149488
- Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004; 2004:re15; http://dx.doi.org/10.1126/stke.2532004re15; PMID: 15467096
- Yu K, Duran C, Qu Z, Cui YY, Hartzell HC. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res 2012; 110:990 - 9; http://dx.doi.org/10.1161/CIRCRESAHA.112.264440; PMID: 22394518
- Sanchez C, Lachaize C, Janody F, Bellon B, Röder L, Euzenat J, et al. Grasping at molecular interactions and genetic networks in Drosophila melanogaster using FlyNets, an Internet database. Nucleic Acids Res 1999; 27:89 - 94; http://dx.doi.org/10.1093/nar/27.1.89; PMID: 9847149
- Satagopam VP, Theodoropoulou MC, Stampolakis CK, Pavlopoulos GA, Papandreou NC, Bagos PG, et al. GPCRs, G-proteins, effectors and their interactions: human-gpDB, a database employing visualization tools and data integration techniques. Database (Oxford) 2010; 2010:baq019; http://dx.doi.org/10.1093/database/baq019; PMID: 20689020
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 2010; 107:14950 - 7; http://dx.doi.org/10.1073/pnas.1005940107; PMID: 20668236
- Miller JP, Lo RS, Ben-Hur A, Desmarais C, Stagljar I, Noble WS, et al. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci U S A 2005; 102:12123 - 8; http://dx.doi.org/10.1073/pnas.0505482102; PMID: 16093310
- Brito GC, Andrews DW. Removing bias against membrane proteins in interaction networks. BMC Syst Biol 2011; 5:169; http://dx.doi.org/10.1186/1752-0509-5-169; PMID: 22011625
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, et al. An in vivo map of the yeast protein interactome. Science 2008; 320:1465 - 70; http://dx.doi.org/10.1126/science.1153878; PMID: 18467557
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. The genetic landscape of a cell. Science 2010; 327:425 - 31; http://dx.doi.org/10.1126/science.1180823; PMID: 20093466
- Babu M, Vlasblom J, Pu S, Guo X, Graham C, Bean BD, et al. Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae. Nature 2012; 489:585 - 9; http://dx.doi.org/10.1038/nature11354; PMID: 22940862
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008; 455:1210 - 5; http://dx.doi.org/10.1038/nature07313; PMID: 18724360
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008; 134:1019 - 29; http://dx.doi.org/10.1016/j.cell.2008.09.003; PMID: 18805094
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008; 322:590 - 4; http://dx.doi.org/10.1126/science.1163518; PMID: 18772398
- Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J Physiol 2009; 587:2127 - 39; http://dx.doi.org/10.1113/jphysiol.2008.163709; PMID: 19015192
- Kunzelmann K, Tian Y, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, et al. Anoctamins. Pflugers Arch 2011; 462:195 - 208; http://dx.doi.org/10.1007/s00424-011-0975-9; PMID: 21607626
- Romanenko VG, Catalán MA, Brown DA, Putzier I, Hartzell HC, Marmorstein AD, et al. Tmem16A encodes the Ca2+-activated Cl- channel in mouse submandibular salivary gland acinar cells. J Biol Chem 2010; 285:12990 - 3001; http://dx.doi.org/10.1074/jbc.M109.068544; PMID: 20177062
- Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, et al. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci U S A 2009; 106:21413 - 8; http://dx.doi.org/10.1073/pnas.0911935106; PMID: 19965375
- Lentz TL. Cell Fine Structure Saunders (W.B.) Co Ltd 1971.
- Duran C, Hartzell HC. Physiological roles and diseases of Tmem16/Anoctamin proteins: are they all chloride channels?. Acta Pharmacol Sin 2011; 32:685 - 92; http://dx.doi.org/10.1038/aps.2011.48; PMID: 21642943
- Tarran R, Loewen ME, Paradiso AM, Olsen JC, Gray MA, Argent BE, et al. Regulation of murine airway surface liquid volume by CFTR and Ca2+-activated Cl- conductances. J Gen Physiol 2002; 120:407 - 18; http://dx.doi.org/10.1085/jgp.20028599; PMID: 12198094
- Stephan AB, Shum EY, Hirsh S, Cygnar KD, Reisert J, Zhao H. ANO2 is the cilial calcium-activated chloride channel that may mediate olfactory amplification. Proc Natl Acad Sci U S A 2009; 106:11776 - 81; http://dx.doi.org/10.1073/pnas.0903304106; PMID: 19561302
- Hengl T, Kaneko H, Dauner K, Vocke K, Frings S, Möhrlen F. Molecular components of signal amplification in olfactory sensory cilia. Proc Natl Acad Sci U S A 2010; 107:6052 - 7; http://dx.doi.org/10.1073/pnas.0909032107; PMID: 20231443
- Stöhr H, Heisig JB, Benz PM, Schöberl S, Milenkovic VM, Strauss O, et al. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J Neurosci 2009; 29:6809 - 18; http://dx.doi.org/10.1523/JNEUROSCI.5546-08.2009; PMID: 19474308
- Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, et al. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J Clin Invest 2010; 120:1240 - 52; http://dx.doi.org/10.1172/JCI41084; PMID: 20335661
- Pifferi S, Cenedese V, Menini A. Anoctamin 2/TMEM16B: a calcium-activated chloride channel in olfactory transduction. Exp Physiol 2012; 97:193 - 9; PMID: 21890523
- Large WA, Wang Q. Characteristics and physiological role of the Ca(2+)-activated Cl- conductance in smooth muscle. Am J Physiol 1996; 271:C435 - 54; PMID: 8769982
- Duan D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J Physiol 2009; 587:2163 - 77; http://dx.doi.org/10.1113/jphysiol.2008.165860; PMID: 19171656
- Xiang CC, Mezey E, Chen M, Key S, Ma L, Brownstein MJ. Using DSP, a reversible cross-linker, to fix tissue sections for immunostaining, microdissection and expression profiling. Nucleic Acids Res 2004; 32:e185; http://dx.doi.org/10.1093/nar/gnh185; PMID: 15604454
- Gokhale A, Larimore J, Werner E, So L, Moreno-De-Luca A, Lese-Martin C, et al. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J Neurosci 2012; 32:3697 - 711; http://dx.doi.org/10.1523/JNEUROSCI.5640-11.2012; PMID: 22423091
- Salazar G, Zlatic S, Craige B, Peden AA, Pohl J, Faundez V. Hermansky-Pudlak syndrome protein complexes associate with phosphatidylinositol 4-kinase type II alpha in neuronal and non-neuronal cells. J Biol Chem 2009; 284:1790 - 802; http://dx.doi.org/10.1074/jbc.M805991200; PMID: 19010779
- Nesvizhskii AI. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 2012; 12:1639 - 55; http://dx.doi.org/10.1002/pmic.201100537; PMID: 22611043
- Gingras AC, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol 2007; 8:645 - 54; http://dx.doi.org/10.1038/nrm2208; PMID: 17593931
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002; 1:376 - 86; http://dx.doi.org/10.1074/mcp.M200025-MCP200; PMID: 12118079
- Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, Vandermoere F, et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol 2008; 183:223 - 39; http://dx.doi.org/10.1083/jcb.200805092; PMID: 18936248
- Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol 2006; 7:952 - 8; http://dx.doi.org/10.1038/nrm2067; PMID: 17139335
- Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc 2006; 1:2650 - 60; http://dx.doi.org/10.1038/nprot.2006.427; PMID: 17406521
- Zlatic SA, Ryder PV, Salazar G, Faundez V. Isolation of labile multi-protein complexes by in vivo controlled cellular cross-linking and immuno-magnetic affinity chromatography. J Vis Exp 2010; 1855; PMID: 20216526
- Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 2010; 11:276 - 87; http://dx.doi.org/10.1038/nrm2866; PMID: 20308985
- Tsukita S, Yonemura S. ERM (ezrin/radixin/moesin) family: from cytoskeleton to signal transduction. Curr Opin Cell Biol 1997; 9:70 - 5; http://dx.doi.org/10.1016/S0955-0674(97)80154-8; PMID: 9013673
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, et al, International Inflammatory Bowel Disease Genetics Constortium. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet 2011; 7:e1001273; http://dx.doi.org/10.1371/journal.pgen.1001273; PMID: 21249183
- Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature 2001; 411:41 - 2; http://dx.doi.org/10.1038/35075138; PMID: 11333967
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005; 122:957 - 68; http://dx.doi.org/10.1016/j.cell.2005.08.029; PMID: 16169070
- Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, Collins BM, et al. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 2010; 141:1220 - 9; http://dx.doi.org/10.1016/j.cell.2010.05.006; PMID: 20603002
- Höning S, Ricotta D, Krauss M, Späte K, Spolaore B, Motley A, et al. Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol Cell 2005; 18:519 - 31; http://dx.doi.org/10.1016/j.molcel.2005.04.019; PMID: 15916959
- Lomant AJ, Fairbanks G. Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J Mol Biol 1976; 104:243 - 61; http://dx.doi.org/10.1016/0022-2836(76)90011-5; PMID: 957432
- Craige B, Salazar G, Faundez V. Phosphatidylinositol-4-kinase type II alpha contains an AP-3-sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell 2008; 19:1415 - 26; http://dx.doi.org/10.1091/mbc.E07-12-1239; PMID: 18256276
- Benashski SE, King SM. Investigation of protein-protein interactions within flagellar dynein using homobifunctional and zero-length crosslinking reagents. Methods 2000; 22:365 - 71; http://dx.doi.org/10.1006/meth.2000.1088; PMID: 11133242
- Huotari J, Helenius A. Endosome maturation. EMBO J 2011; 30:3481 - 500; http://dx.doi.org/10.1038/emboj.2011.286; PMID: 21878991
- van Meel E, Klumperman J. Imaging and imagination: understanding the endo-lysosomal system. Histochem Cell Biol 2008; 129:253 - 66; http://dx.doi.org/10.1007/s00418-008-0384-0; PMID: 18274773
- Galli T, Chilcote T, Mundigl O, Binz T, Niemann H, De Camilli P. Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J Cell Biol 1994; 125:1015 - 24; http://dx.doi.org/10.1083/jcb.125.5.1015; PMID: 8195285
- Veale KJ, Offenhäuser C, Whittaker SP, Estrella RP, Murray RZ. Recycling endosome membrane incorporation into the leading edge regulates lamellipodia formation and macrophage migration. Traffic 2010; 11:1370 - 9; http://dx.doi.org/10.1111/j.1600-0854.2010.01094.x; PMID: 20604897