Abstract
The use of fluorescence microscopy is central to cell biology in general, and essential to many fields (e.g., membrane traffic) that rely upon it to identify cellular locations of molecules under study and the extent to which they co-localize with others. Rigorous localization or co-localization data require quantitative image analyses that can vary widely between fields and laboratories. While most published data use two-dimensional images, there is an increasing appreciation for the advantages of collecting three-dimensional data sets. These include the ability to evaluate the entire cell and avoidance of focal plane bias. This is particularly important when imaging and quantifying changes in organelles with irregular borders and which vary in appearance between cells in a population, e.g., the Golgi. We describe a method developed for quantifying changes in signal intensity of one protein within any three-dimensional structure, defined by the presence of a different marker. We use as examples of this method the quantification of adaptor recruitment to transmembrane protein cargos at the Golgi though it can be directly applied to any site in the cell. Together, these advantages facilitate rigorous statistical testing of differences between conditions, despite variations in organelle structure, and we believe that this method of quantification of fluorescence data can be productively applied to a wide array of experimental questions.
Introduction
The use of fluorescence microscopy has grown increasingly common throughout the biological sciences with continuous improvements in sensitivity, resolution, and applications. Fluorescence-based microscopy is routinely used to answer questions about where in a cell a given protein can be found, which organelle(s) it transits through, which other proteins are found in the same locations or in the same traffic pattern. These questions are increasingly important to cell signaling research, in which soluble proteins may be recruited to membrane surfaces, often in a highly regulated and transient fashion. In such cases the percentage of one protein co-localizing with another can change dramatically and rapidly. Co-localization, or the quantification of the amount of overlap between two fluorophores, within two dimensional dual-colored images has been an important and powerful tool in determining a protein’s location and its potential for acting at specific sites or with specific binding partners. Localization of a protein to discrete structures (e.g., centrosomes, nuclear pores and ER exit sites) can provide striking evidence of co-localization with markers of those structures (γ-tubulin, lamin B and Sec13, respectively) that often do not require statistical analyses. In contrast, other structures (e.g., the Golgi and endosomes) are irregular in morphology, vary in their three dimensional structures across individual cells within a population and contain within them multiple overlapping domains that require more sophisticated statistical analyses. This variability in organelle morphology and within the sampled population also introduces a much greater potential for focal plane bias that can unintentionally skew the later analyses or images used to portray results. These concerns are ameliorated by the collection of data from the entire cell and multiple cells within the population but require specific statistical analyses to draw conclusions regarding differences between conditions in the experimental design. How the imaging data are collected and processed is obviously critical to those analyses.
All images contain both in focus and out of focus light, potentially confounding quantitative analysis. Confocal microscopy, in which the use of a laser light source and pinhole apertures filter unwanted light from the sample (for reviews, see refs. Citation1 and Citation2) can greatly reduce the amount of out of focus light in an image. But confocal microscopy requires a relatively strong signal, can suffer from rapid photobleaching of the sample, and can require multiple passes (averaging) over the sample to obtain the image.Citation1 Alternatively, widefield microscopy illuminates the entire sample simultaneously (with a lower energy light source) and the image is acquired using a charged coupled device (CCD) camera, allowing for less photobleaching and much more rapid data acquisition.Citation2 However, the resultant image contains light originating from throughout the entire sample, i.e., out of focus light is present in each slice of the stack. Algorithms have been developed to deconvolve the image, to remove out of focus light and reassign it to its point of origin (for review, see ref. Citation3). This process can be applied to two dimensional images, or stacks of images collected on either confocal or widefield systems.Citation4 For detailed reviews of deconvolution see references Citation3, Citation5 and Citation6.
Once images are acquired and deconvolved, most methods of quantification require the images to be thresholded, or processed to discard noise, which includes low intensity, background signal, or signal contributed by the electronics within the system itself (often referred to as Poisson noise).Citation7 Thresholding effectively removes noise by disregarding pixels with intensities that fall below a specified value, often determined by visually inspecting the intensity profile or a histogram of intensities within the image. Thresholding can affect the outcome of the analysis by inadvertently removing low intensity signal that should have been included. While the method of thresholding should always be carefully considered it is particularly important when the source of the signal under study is present in both diffuse, soluble (cytoplasmic) and particulate, membrane-associated forms, such as proteins (adaptors, kinases, lipid modifiers, etc.) whose binding to membranes is transient and regulated. Images of this nature often benefit from statistical analysis of a collection of images because of the potential for thresholding-induced bias, the irregular, three dimensional structure being observed, the large area over which the protein of interest may be recruited and the reversible nature of the interactions.
Co-localization, or quantification of the amount that two fluorophores correlate or overlap, is commonly used to evaluate location when one is a previously characterized marker of an organelle/structure. There are several ways to statistically analyze co-localization; including, but not limited to, Pearson’s correlation coefficient (PCC) and Mander’s overlap coefficient (MOC). These measures are commonly applied to confocal images, and work very well with fluorophores of comparable, strong pixel intensities. However, co-localization methods require thresholding and well-defined regions of interest (ROI), the more tightly defined the ROI the better the analysis. Additionally, the output of a co-localization analysis yields a single, correlative value for each image that contains far less information than is available from using other methods. Further statistical analyses of multiple images and their corresponding co-localization scores produce a mean correlative score from which one can conclude the extent of overlap. In contrast, other methods make use of the information available in three dimensional data sets and allow users to extract more spatial information, including numbers of objects, relationships among objects (clustered vs. dispersed, etc.), and regional differences within objects based upon intensity.Citation8,Citation9
We wanted to be able to generate irregularly shaped three dimensional ROIs that are defined by the presence of a marker of a specific cellular compartment and evaluate pixel intensities within those volumes, or isosurfaces. Isosurfaces are computational, three dimensional representations of a structure within an image stack and can be generated using a number of methods that are designed to detect boundaries between regions of signal and non-signal. Such methods include watershed segmentation, gradient detection, or iterative selection.
Watershed segmentation uses a theory first described 100 years agoCitation10 and imagines water falling onto or flooding from the bottom of a topological representation of pixel intensities within an image to determine where water would pool on the topology map. It then defines objects by drawing borders around areas from which the theoretical water was shed.Citation11 The user can set the amount of theoretical water allowed to fall in the system, and determine the amount of “land” identified as “object” in the image. Gradient detection calculates intensity differences between two pixels and uses that information to identify objects. Contiguous pixels with small intensity differences that are above a certain user-defined threshold define an object. Software packages (e.g., Imaris Bitplane Scientific Software) use watershed and gradient detection as methods of identifying objects. Some methods use combinations of these approaches to automatically identify and count objects, such as the freely available ImageJ plug-in “WatershedCounting3D.”Citation12 WatershedCounting3D is very good at detecting areas of local contrast over a wide range of pixel intensities and when dealing with strong signal that is clearly distinct from background. However, this approach employs thresholding, and images that contain diffuse, cytosolic staining can result in the identification of objects in areas that are distinctly background or cytosolic staining.
Finally, iterative pixel selection, or the Calvard method,Citation13 and its variations, evaluates an image a number of times (Imaris software default is four times), each time scoring pixels as “object” or “non-object.” It calculates two populations of pixel intensities, maximizing the statistical differences between them and defines pixels within the higher population to be those containing signal that corresponds to an object, and the lower population to comprise noise. It then generates a map, or mask, of pixels that were identified as object and applies it to the original image. The process is then repeated on pixels that fall into the region defined by the mask. Each iteration results in a more refined object, each time optimizing the statistical difference between the two populations to result in the largest possible difference between foreground (object) and background (non-object). This approach is advantageous because it (a) retains all information in the data set, i.e., does not throw out data below any limit, (b) does not require thresholding prior to object identification and therefore evaluates all pixel intensities within the raw image, (c) is dynamic in that it can be applied equivalently to images that vary in their intensity profiles (may be dimmer or brighter than other images in a data set, or have more background, etc.) and (d) can be applied automatically to all images included in an analysis. In a comparison of multiple methods of object identification, iterative selection was the only approach that was determined to be a suitable replacement for manual inspection.Citation14
We present a method specifically designed for the quantification of three dimensional data sets that identifies volumetric structures and evaluates signal intensity information within those structures using three dimensional (3D) image-based isosurface generation and intensity analysis (3D3I). We believe this method provides a useful addition to imaging data analyses with the specific advantages of (a) automatic object identification that can be applied easily to many images of comparable or disparate intensity profiles (i.e., applicable to strong localized signal or diffuse cytosolic staining), (b) eliminating the need for thresholding, (c) generating real values rather than correlative ones, (d) allowing for the statistical detection of outliers based on the characteristics of the image and (e) being particularly well suited for analyses of protein recruitment to membranes but can be applied to other changes in protein distribution in cells.
Results
Procedure for 3D3I image analysis
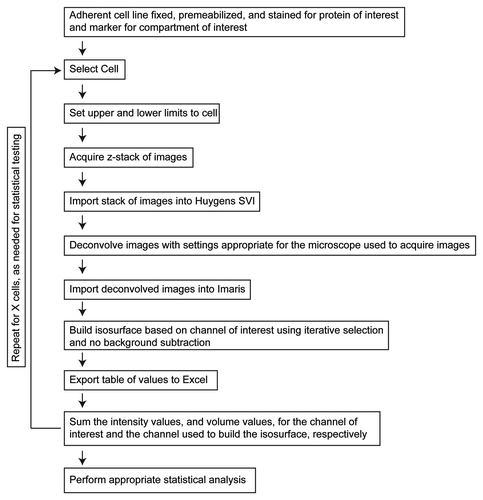
Z-stacks of wide field images using a 60× objective were collected for two channels from the entire volume of the cell using a step size of 0.2 μm, as described under Materials and Methods. Exposure times of 200 ms were used for each channel. This method can accommodate the use of either confocal or wide field imaging systems, however we chose to use wide field to minimize problems resulting from photobleaching. The acquired stack of images was then deconvolved to remove out of focus light. The deconvolved stacks were imported into Imaris. The channel used to identify a cellular structure (e.g., Golgi marker) was automatically segmented using the iterative selection method as implemented with Imaris software, and an isosurface, or computational volume that represents the intensities within the channel used to generate it, was generated. Intensity values for both channels within the isosurface were then exported to Excel. This output includes a list of each isosurface identified in the image, their volumes, and intensities of signal in each. highlights the workflow for quantification of isosurfaces from cells.
Figure 1. 3D3I Workflow. Steps in the performance of 3D3I to quantify staining in fixed adherent cells in culture are illustrated. We describe throughout the use of one well defined organelle marker (e.g., giantin, TGN46, etc.) to define the isosurface into which the other antigen is compared but any two antigens can be compared in the same way. Wide field images were collected in z-series throughout the volume of the cell to create an image stack. This stack is imported into Huygens SVI software and deconvolved to remove out of focus light. The deconvolved stack is imported into Imaris and an isosurface is built, based upon staining of the organelle marker. An important aspect of this method is that the isosurface is generated without thresholding the stack in any way. Information about the isosurfaces generated by Imaris is then exported to Excel for use. Each object within the isosurface is assigned an identification number with corresponding volumes, sum channel intensities within each object, maximum intensities per object, intensity mean, intensity minimum, standard deviation of intensity and surface areas. This list of exported values is not comprehensive but representative. The return loop is intended to show that the process can be repeated on as many cells as required for the statistical tests used.
Problems arising from the collection and analysis of three dimensional data sets
The ability to collect images at different z-positions throughout the volume of the cell allows the generation of three dimensional reconstructions that provide much more information than that present in a single two dimensional image but raises an important issue that is worthy of specific elaboration. The analysis of one image from a single plane of focus can result in unintentionally biased results, termed focal plane selection bias. Collecting and using data from an entire cell volume should effectively eliminate such bias. This is apparent when two spatially separated images collected from the same cell are evaluated for co-localization. Co-localization methods can evaluate how well two channels correlate in their intensities (e.g., bright pixels on one channel coincide with bright pixels on the other), or how well two channels overlap (e.g., where there is signal on one channel there is signal on another).
The classical measure of co-localization is the Pearson’s Correlation Coefficient (PCC) and is the origin for most co-localization measures used today.Citation15,Citation16 A PCC value of -1 would be interpreted as inverse correlation (where pixels in one channel are brighter than the mean, pixels in the other channel are likely to be dimmer than the mean pixel intensity) while a PCC value of 0 would indicates no correlation (there is no relationship between the two channels), and a PCC value of +1 indicates perfect correlation (where pixels in one channel are brighter than the mean, pixels in the other channel are also brighter than the mean). While the PCC indicates how well or how poorly two channels vary with one another, some questions are better answered with information about the degree to which two channels overlap, rather than how well their intensities co-vary. To obtain an equation that monitors overlap of pixels, the PCC is modified to evaluate absolute pixel intensity rather than deviation from the mean intensity, and results in the Mander’s Overlap Coefficient (MOC).Citation16,Citation17 Because the MOC evaluates absolute intensities rather than the co-variance of two channels it eliminates the possibility of a negative correlation. The MOC can be further rearranged to give two scores, one for each channel, rather than one score for an entire image. These coefficients are referred to as M1 and M2 and indicate how much “red signal is also green,” or vice versa.
To illustrate these issues, PCC and MOC measures were calculated for two slices isolated from a single stack of deconvolved wide field images to evaluate the degree of co-localization in two spatially distinct regions of the same cell. Stacks of images were collected from fixed HeLaM cells expressing a protein containing the lumenal and transmembrane domains of CD8 fused to the cytoplasmic tail of furin (CD8-furin), which (like furin itself) localizes predominantly to the Golgi.Citation18,Citation19 Fixed cells were labeled with antibodies directed against TGN46, a marker of the trans-Golgi network (TGN) compartment (, red), and to CD8, to detect the cargo (, green). Stacks were deconvolved with Huygens SVI software, the two channels were merged and a montage was generated (). The sixth and the sixteenth images were arbitrarily selected, extracted from the stack and evaluated for co-localization using the “Co-localization Threshold” plug-in (http://pacific.mpi-cbg.de/wiki/index.php/Colocalization_Threshold) from FiJi (). This plug-in calculates the PCC as well as the MOC and thresholded M1, M2 (tM1, tM2) scores for each channel. The PCC and tM1/M2 measures were calculated for the two slices and are shown in , along with a scattergram of overlapping intensities where the colder/darker pixels represent less frequent occurrences of a given pixel intensity, while warmer/yellow colors indicate a higher frequency of occurrence. The amount of co-localization, as determined by either PCC or tM, differs quite a bit between the two, spatially distinct slices. This variation results from different focal planes will certainly add to the variation between samples, e.g., when averaging a correlative score among different cells from a single population, and can even alter conclusions drawn from the data set. Allowing the user to select by visual inspection one image from the z-stack for comparison to other cells increases the potential for introducing unintentional bias. The use of three dimensional data sets avoids focal plane bias by using data from throughout the cell but require optimized methods for analysis.
Figure 2. Focal plane bias in use of single images. (A) Stacks of images were collected from HeLaM cells expressing CD8-furin and stained for both CD8 and TGN46, as described under Materials and Methods. Z-stacks (one for each channel) were then deconvolved, the deconvolved stacks opened in ImageJ, merged and a montage created that shows 18 individual images at different depths in the z-plane. (B) Choice of focal plane can affect results of quantification of co-localization. The sixth and the sixteenth slices within the z-stack were arbitrarily selected for co-localization analysis. The two slices were evaluated for PCC, and thresholded Mander’s coefficients (tM1 and tM2) and intensity histograms generated.
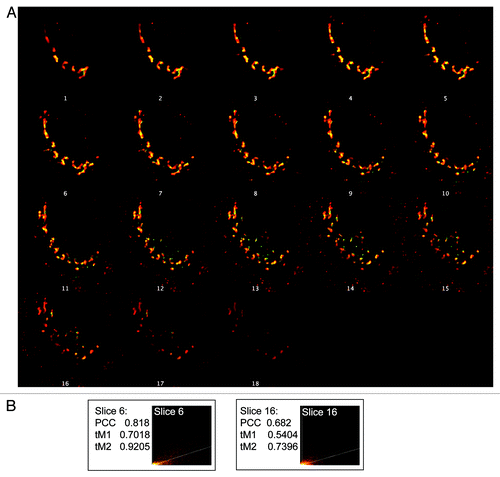
Image segmentation using WatershedCounting3D and 3D3I
We compared objects identified using 3D3I analysis to those identified using a similar method, watershed segmentation.Citation12 Using the same stack of images shown in in which cells expressing CD8-furin were fixed and stained for TGN46, a marker of the trans-Golgi network and CD8, we compared the regions identified as objects/structures using watershed segmentation to those identified using 3D3I. The images used have a linear range of pixel intensities as shown for the TGN46 marker in (CD8 staining intensities not shown), with no easily identifiable, discrete populations of pixel intensities, making them difficult to threshold manually. The merged CD8 and TGN46 stacks were mapped onto a three dimensional intensity plot to represent intensities of the two channels (). Note the gray, textured regions of low intensity, illustrating variations in background signal, and corresponding to low-intensity pixels seen in the histogram shown in . WatershedCounting3D segmentation was applied to the TGN46 channel and a mask of the segmented regions was generatedCitation12 (). We believe that (perhaps because there is a linear range of pixel intensities) the Watershed method identifies a large number of discrete isosurfaces; far more than would be expected for the biological structures that contain TGN46. This conclusion was further supported by the observation that the staining seen in the upper right quadrant of the images shown in is coming from the nucleus, which is expected to lack TGN46. The mask was then falsely colored blue, and applied to the three dimensional intensity plot (). Comparing to , ( is the addition of and ) it is clear that while the WatershedCounting3D algorithm is excellent at detecting areas of local contrast over a wide range of pixel intensities, its ability to detect areas of local contrast resulted in the identification of objects containing very low-intensity pixels (, arrows). These low-intensity objects can be removed by thresholding the image prior to analysis. However, to do so we must manually assign a cutoff intensity that may or may not be applicable over large numbers of image stacks. In comparison, we generated an isosurface based on TGN46 signal using the iterative selection method implemented in Imaris (, green) and compared it with the TGN46 channel (, red). It is evident by simple inspection that the isosurfaces generated using the iterative selection method (, green) faithfully capture the overwhelming majority of the staining seen in the primary data (, red).
Figure 3. Comparison of image segmentation using WatershedCounting3D and 3D3I. The same HeLaM cell, expressing CD8-furin and stained for CD8 and TGN46, shown in was used for analyses. (A) Histogram of intensities from a z-stack of deconvolved images of TGN46 staining; the maximum intensity projection is shown in the inset. (B) The three dimensional surface plot of pixel intensities for TGN46 (red) and CD8 (green) is shown as the merged image. Height indicates relative intensity. (C) WatershedCounting3D was used to identify objects and create a mask based on TGN46 staining. (D) The mask shown in (C) was false-colored blue and imported into the three dimensional intensity plot shown in (B). Blue regions indicate areas contained in the segmented mask. Arrows highlight regions of the mask that occur in areas of low intensity. (E) We used the same deconvolved z-stack of TGN46 (red) staining to generate an isosurface using Imaris (shown in green) as described under Materials and Methods. Isosurface generation results in fewer objects being identified in the regions of low signal intensity [e.g., compare panel (E) to (C)].
![Figure 3. Comparison of image segmentation using WatershedCounting3D and 3D3I. The same HeLaM cell, expressing CD8-furin and stained for CD8 and TGN46, shown in Figure 2 was used for analyses. (A) Histogram of intensities from a z-stack of deconvolved images of TGN46 staining; the maximum intensity projection is shown in the inset. (B) The three dimensional surface plot of pixel intensities for TGN46 (red) and CD8 (green) is shown as the merged image. Height indicates relative intensity. (C) WatershedCounting3D was used to identify objects and create a mask based on TGN46 staining. (D) The mask shown in (C) was false-colored blue and imported into the three dimensional intensity plot shown in (B). Blue regions indicate areas contained in the segmented mask. Arrows highlight regions of the mask that occur in areas of low intensity. (E) We used the same deconvolved z-stack of TGN46 (red) staining to generate an isosurface using Imaris (shown in green) as described under Materials and Methods. Isosurface generation results in fewer objects being identified in the regions of low signal intensity [e.g., compare panel (E) to (C)].](/cms/asset/802d51ac-cdd6-4ae4-b80c-c28cb9f84325/kcll_a_10923150_f0003.gif)
A comparison of the isosurface in to the blue regions identified in revealed that far fewer objects (discrete isosurfaces) were identified using the Imaris isosurface generation approach than in watershed segmentation. Due to file format incompatibility we were unable to project the Imaris isosurface onto the ImageJ three dimensional intensity space. Given our current understanding of the TGN compartment we believe that many of the low peaks seen in (e.g., arrows) do not faithfully identify the TGN. Because the iterative selection method maximizes the statistical difference between signal and non-signal pixels, and effectively utilizes the dynamic range of pixel intensities within an image, the overall intensity of a signal is less of an issue in identifying isosurfaces. In addition, in our hands each image evaluated using WatshedCounting3D required continuous adjustments to the settings to filter out the objects identified in regions of low signal intensity. We also noted that two dimensional confocal images resulted in better segmentation using the WatershedCounting3D method but the use of three dimensional data sets, and the individual settings necessary to analyze each image, make this method prohibitive to the analysis of large numbers of images. For studies that will use statistical analyses comparing different conditions or data sets, a method is required that involves as little image processing as possible, and yet is robust enough to be applicable over a wide range of signal profiles. In summary, a direct comparison of 3D3I to watershed counting reveals that fewer objects are identified in regions of low contrast (refer to ) using 3D3I and, at least in the case of this marker of the TGN, give us higher confidence that we are quantifying the biologically relevant compartment under study. Further testing and comparisons should be performed to determine if this holds up over a wider array of organelle markers; though we have used this method for quantification of transferrin receptor containing vesicles (recycling endosomes), early endosomes, and lysosomes, with similar results to those obtained for Golgi proteins.
Development of the 3D3I method for quantifying recruitment of adaptors to irregular, three-dimensional structures, such as the Golgi and endosomes
Because the Golgi and endosomes are irregularly shaped structures that can appear quite different between cells and in different focal planes of the same cell, we sought a method that is inclusive of all staining in each cell used for quantification. A related issue is that diffuse, cytosolic staining can be difficult to appropriately threshold and depending upon how it is handled, can result in exaggerated or understated co-localization scores (see below). To address these issues, we wanted a method that would allow for the identification of three dimensional objects without discarding cytosolic staining. To this end, we used wide field imaging with deconvolution followed by analyses using Imaris software, as described under Materials and Methods. Organelle marker staining (e.g., giantin for the Golgi) was used to generate isosurfaces and the staining of the adaptor Mint3 was determined within those isosurfaces. Note that this method allows for the identification of multiple, discrete objects within the same cell and outputs include numbers and volumes of all objects (see ). Though many parameters are calculated for each isosurface, we chose to score images as a ratio of total pixel intensity of adaptor per unit volume of isosurface (defined by the marker of the Golgi) from each cell in the units, intensity/μm3. We chose this metric (intensity/μm3) because it best addressed the question “how much Mint3 is on the Golgi?” and allowed us to compare the answers from different cell populations in a statistically rigorous fashion. However, the 3D3I method can be tailored to evaluate other questions that are better described using other metrics. For example, if one is interested in fragmentation 3D3I can address that simply by arranging the values that are exported to best describe the question. The isosurface serves as a tightly defined three dimensional ROI, or volume within which we wish to monitor changes. This method has a number of advantages: (1) all data included are defined by a biologically relevant marker used to define the isosurface (avoiding over-estimates resulting from correlational co-localization methods), (2) we monitor changes throughout the entire volume of the cell and thus avoid sampling or focal plane bias and (3) we perform statistical analyses on a number of cells, comparing intensity per unit volume in control vs. experimental conditions, as opposed to performing statistical analysis on mean correlational scores. Importantly, because all data are included, including low intensity signal throughout the cell, we typically observe smaller fold-changes in response to experimental manipulations using 3D3I than those found in more commonly used co-localization methods.
An example of the use of 3D3I to quantify adaptor recruitment is shown in , with comparison to co-localization methods. Wide field images of control HeLaM cells were collected and deconvolved using Huygens’ Deconvolution software (SVI Inc.). Deconvolved images were opened in Imaris and analyzed for co-localization using automatic thresholding. A co-localization mask, or a graphical representation of pixels that overlap, was generated (, “Co-localization Mask”). Imaris calculated a Pearson’s coefficient of 0.5211, as well as thresholded Mander’s coefficients of 0.7605 and 0.3871 for giantin and Mint3, respectively (). These coefficients were calculated for the entire stack though the images shown are maximum intensity projections. We then performed the same type of analysis using ImageJ and compared the results to those obtained using the colocalization calculator in Imaris. The same deconvolved images were opened in ImageJ and converted to 16-bit images, which were used to generate the values in , and a frequency plot of intensities () using the “Mander’s Calculator” plug-in from the MacMaster Biophotonics Facility (www.macbiophotonics.ca/imagej). The overall shape of the scattergram indicates a positive correlation between giantin and Mint3 pixels. It is clear that there are more Mint3 pixels that do not have giantin signal in them (see the clustering of warm colors along the y-axis) than giantin pixels devoid of Mint3 intensity as indicated by the absence of a similar lobe of intense pixels along the x-axis. That is, most giantin positive pixels are also positive for Mint3 but not all Mint3 positive pixels are positive for giantin. Such incompleteness in overlap is not surprising, given the complex organization of the Golgi itself and the extensive documentation that markers of this organelle display regions of non-overlap (i.e., giantin stains a portion of the Golgi but not all of it, and Mint3 is present on surfaces of the Golgi as well as endosomes). We interpret this partial overlap as evidence of Mint3 recruitment to the Golgi, including some areas that are giantin positive and some that are not. The Pearson’s coefficient generated using the ImageJ software is similar to that obtained from Imaris. Mander’s coefficients obtained from both methods determined that most of the giantin intensity was also positive for Mint3 (compare the Imaris value of 0.7605 to the ImageJ value of 0.979) while there were fewer Mint3 positive pixels that were also positive for giantin (compare the Imaris value of 0.3871 to the ImageJ value of 0.678). The differences between values generated by ImageJ and Imaris most likely arise from the manner in which each software package handles thresholding. Though neither value is more correct than the other, it suggests that thresholding be approached cautiously, and used sparingly. However, both methods generate the same rank order of tM coefficients in that giantin is higher than Mint3 under each treatment. In summary, the manner in which co-localization calculations are implemented within a given software package can influence substantially the quantification of an image.
Figure 4. Comparison of 3D3I to other co-localization methods. HeLaM cells were fixed and labeled with antibodies against giantin and Mint3. (A) Images were collected using widefield imaging and a step size of 0.2 μm. Stacks were deconvolved using Huygens deconvolution software (left two panels) and Imaris was used to generate the co-localization mask of the two channels (third panel), as described under Materials and Methods. A merge of the two channels and the colocalization mask are shown (fourth panel). Maximum intensity projections are shown. (B) Pearson’s and thresholded Mander’s (tM) coefficients were calculated using Imaris and ImageJ, as described under Materials and Methods. (C) Imaris was used to generate a scatter plot of pixel intensities using the merged giantin and Mint3 channels shown in (A). (D) The same deconvolved images shown in (A) (left two panels) were opened in Imaris and merged (third panel). The giantin channel (red) was used to create the isosurface (right panel). (E) The isosurface generated in panel (D) is color-coded as a heat map of Mint3 intensities contained within each object in the isosurface. Representative values describing the isosurface are shown in the panel to the right of the color-coded isosurface. Note the variations in Mint3 intensity within the giantin isosurface, based on the color-coding shown. (F) The co-localization mask shown in (A) was falsely-colored green, and the isosurface generated in (D) was falsely-colored red. The two are displayed simultaneously in the merged image. The isosurface is a more tightly defined volume as the two representations overlap, but the co-localization mask highlights regions not identified by the isosurface.
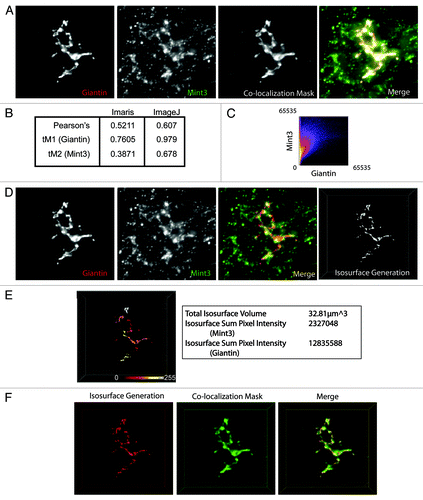
We wanted a method to generate values containing absolute intensity information about a given channel within a defined three-dimensional region of interest as opposed to an entire image or cell. To do this, the same images used in were analyzed with the Imaris software package using the iterative selection method to automatically define an isosurface that was representative of all giantin staining (). From this surface, a heat map of Mint3 intensities was generated to illustrate the point that not all giantin staining has a uniform amount of Mint3 staining. Instead there are regions of the isosurface that have high (white) or low (dark red) levels of Mint3 intensity (). This level of spatial detail is lost when the image is evaluated using co-localization, or statistical correlation methods. Data describing the isosurface, such as total isosurface volume, and the pixel intensities of Mint3 and giantin contained within that volume are shown (, right). When expressed as a ratio of sum intensity per isosurface volume, the information can be used to make comparisons across many groups and are suitable for statistical analysis. We then wanted to compare the region of pixels that were determined to co-localize with the region that was defined by the isosurface (). The isosurface (red) and the co-localization mask (green) were displayed simultaneously in Imaris (merge), and exported as a .tif file. From the merged image it is clear that there are regions defined by the co-localization mask that do not also contain regions defined by the isosurface. However, all of the isosurface volume contains volumes defined by the co-localization mask.
3D3I Applications
3D3I analysis can be used to analyze data from many different types of experiments and is particularly well suited to making comparisons across multiple conditions. To illustrate this point, 3D3I was used to analyze Mint3 recruitment to isosurfaces defined by the presence of the amyloid precursor protein (APP; ). The cytosolic tail of APP interacts with the adaptor protein Mint3 at the Golgi for packaging and export.Citation20-Citation22 APP is a transmembrane protein that transits both the secretory and endocytic pathways and is proteolytically processed in the process. This can lead to difficulties in interpretation when using N- or C-terminal directed antibodies or tags on APP as they may become separated as a result of protease activities. For this reason, we compared our results of expressed wild type human APP to those of a recombinant protein consisting of the lumenal and transmembrane domain of CD8 and the cytoplasmic tail of APP (CD8-APP) that is completely comparable to the CD8-furin construct used above. Use of the CD8-APP construct also allows us to monitor traffic of a protein independently of lumenal domain interactions, which have been described previously for APP.Citation23 We obtained the same results whether full length APP or CD8-APP was expressed so examples of each are presented in . We also used the previously characterized temperature block protocol, in which cells were maintained at 20°C for 4 h, during which time protein synthesis and export from the ER continued (albeit at slower rates than if cells are cultured at 37°C) but export from the Golgi/TGN was compromised.Citation24-Citation27 HeLaM cells were transfected with a plasmid directing expression of human APP695 () or CD8-APP () and the next day were either maintained at 37°C or the temperature block was imposed. Cells were then fixed and stained with the antibodies indicated, as described under Materials and Methods. Stacks of wide field images were collected and deconvolved using Huygens SVI software. Deconvolved images were opened with Imaris and isosurfaces were generated based on APP staining () or based on giantin staining (). The amount of Mint3 staining within those isosurfaces was compared between conditions.
Figure 5. Example of a 3D3I application. Temperature block increases the recruitment of Mint3 to APP at the Golgi. HeLaM cells were transfected with empty plasmid or ones directing expression of full length human APP (A), (B) or CD8-APP (D) and the next day were fixed with or without imposition of the temperature blockade, as described under Materials and Methods. Cells were then stained for (A) APP and Mint3 or (B) Mint3 and giantin. Maximum intensity projections of widefield images are shown with isosurface generation using APP (A) or giantin (B) staining. Heat maps indicate the intensity of Mint3 staining within those isosurfaces. (C) The amount of Mint3 staining per APP volume was determined under the conditions shown in panel (A). (D) Control HeLaM cells, or cells expressing CD8-APP were maintained 37°C or temperature blocked prior to fixation, and stained with antibodies directed against Mint3 or CD8. Bars in (C and D) show the average from n ≥ 7 cells per condition, representative of at least three independent experiments. Error bars indicate standard error of the mean (SEM). Asterisks indicate a p < 0.01, each compared with control, steady-state staining.
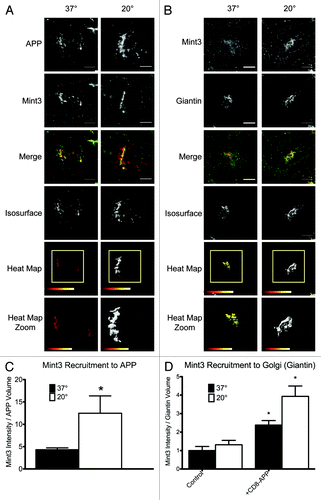
The increased recruitment of Mint3 to APP isosurfaces in response to imposition of the temperature block is easily seen in the heat map at the bottom of and quantified in . We quantified the amount of Mint3 signal within isosurfaces defined by APP staining and compared the results obtained from cells maintained at 37°C and 20°C (). Sum intensity values were expressed as the ratio of Mint3 per isosurface volume and the data from at least seven cells were averaged and used in statistical analyses. Results demonstrated that the temperature block led to a statistically significant increase (p < 0.01) in Mint3 within APP isosurfaces.
We predicted that the temperature blockade would lead to an increase in APP at the Golgi due to the decrease in anterograde traffic from the Golgi under this condition and asked whether the Mint3 adaptor was being recruited to that site. Thus, in a parallel set of experiments Mint3 recruitment to the Golgi (defined by giantin staining) was quantified in control cells and compared with those expressing CD8-APP, with or without temperature block (). It is evident from the bar graph () that imposition of the temperature block alone does not cause an increase in Mint3 at the Golgi. In contrast, expression of CD8-APP does increase Mint3 at the Golgi and this effect is further increased upon imposition of the temperature block.
We evaluated sum intensity within an isosurface in these examples, but many other parameters can be measured using 3D3I; including isosurface volume, number of isosurfaces, number of voxels within an isosurface, mean intensity and intensity standard deviation. These parameters are more tangible than the correlative measures generated in co-localization analyses and retain the spatial information present in the original image.
Discussion
We conclude that the use of three dimensional isosurface generation and of intensities contained within that volume (termed 3D image-based isosurface generation and intensity analysis, or 3D3I analysis) generates non-correlative, raw values suitable for statistical analysis of large numbers of cells and confined to marker-defined, physiologically relevant regions or structures. As such we believe this method is a valuable alternative to correlative values when information about a three dimensional structure is of interest. This method is particularly suited to irregularly shaped organelles and ones that can appear so different between cells in the same population, like the Golgi and endosomes, but is flexible enough to be used to address a variety of types of questions that may be relevant to any site or cellular organelle.
Materials and Methods
Cell culture
HeLaM cells were maintained at 37°C and 5% CO2 in 10% fetal bovine serum (GemCell, 100–500) in DMEM medium (GIBCO, 11965). Temperature blockade was performed as previously describedCitation24-Citation27 and involved aspirating off medium from cells grown on coverslips, replacing it with DMEM containing 20 mM HEPES, pH 7.4 with 10% fetal bovine serum, pre-warmed to 19.5°C, and maintaining cells in a water bath at the same temperature for four hours. Cells were then either fixed and processed immediately or allowed to recover by return to 37°C for the times indicated, as described below.
Plasmids and transfections
The pIRESneo parent plasmid used to express the lumenal and transmembrane domain of CD8 fused to the cytoplasmic domain of furin, termed CD8-furin, was a generous gift from Dr. Matthew Seaman (University of Cambridge).Citation28 Expression of full-length human amyloid precursor protein, APP695, was achieved as described in Shrivastava-Ranjan et al.Citation20 We also generated a plasmid encoding the CD8 lumenal and transmembrane domains fused to the cytoplasmic tail of APP, consisting of the 46 C-terminal residues (residues 650–695), termed CD8-APP. The APP tail was inserted into pIRESneo-CD8 using NotI and AflII restriction sites.
Cells were plated at ~80% confluence and the next day were rinsed with pre-warmed, serum free Opti-MEM (GIBCO, 11058) and then placed in 1.0 mL of pre-warmed Opti-MEM. Fugene transfection reagent (6 μL; Roche, 11814443001) was added to OptiMEM (93 μL) in a microfuge tube. DNA (1 μg) was then added to the Opti-MEM/Fugene solution, tapped gently to mix, and incubated at room temperature for 20 min. The transfection mixture was then added drop wise to one well of a 6-well plate and placed at 37°C for 5 h. The transfection was stopped by rinsing cells once with pre-warmed Trypsin-EDTA (GIBCO, 25300) and then adding 0.5 ml 0.05% Trypsin-EDTA and incubating at 37°C for ~5 min. Cells were triturated off the plate using 3.5 mL of pre-warmed growth medium and plated onto 6 cm dishes containing Matrigel (BD Biosciences, 356234)-coated coverslips and allowed to attach overnight.
Immunocytochemistry
Cells plated onto coverslips the night before were fixed with 2% paraformaldehyde diluted in phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate monobasic, pH 7.4) for 20 min at room temperature. Fixative was aspirated off and cells were rinsed four times for 5 min each with PBS. Non-specific staining was blocked by incubating cells in 200 μL of blocking solution [1% bovine serum albumin (Sigma, A3059) and 0.05% saponin (Sigma, S5881)] in PBS for 20 min at room temperature. Antibodies against human TGN46 (Serotec, AHP500, 1:1,000), APP (Synaptic Systems, 127002, 1:500), Mint3 (BD Transduction, 611380, 1:200), or giantin (Covance, prb114c, 1:1,000) were diluted in blocking solution and applied to the cells overnight (~16 h) at 4°C. The following morning, cells were washed four times for 5 min each in 0.05% saponin (Sigma, S5881) in PBS (SAP) at room temperature. Secondary antibodies (Alexa 594 and Alexa 488; Invitrogen, A11016 and A11008, respectively) were diluted 1:500 in blocking solution and applied to the cells for one hour at room temperature, protected from light. Cells were then washed twice in SAP, 5 min each. When indicated, FITC-conjugated anti-human CD8 (Ancell Corp., 153-040) was diluted 1:1,000 in blocking solution and ~150 μL was added for 1 h at room temperature, protected from light. Coverslips were then washed two times 5 min each in SAP. Hoechst dye was diluted 1:5,000 in blocking solution and applied to cells for 5 min. Cells were washed twice more in SAP, rinsed once in PBS, 5 min each. Coverslips were mounted onto slides using Mowiol (CalBiochem, 475904), prepared as described in Valnes and Brandtzaeg.Citation29
Wide field image acquisition and deconvolution
Stacks of images were collected using a Nikon TE300 microscope with a 60× 1.4NA oil immersion objective with a Photometrics Quantix camera. Cells were randomly selected for imaging if they (1) expressed CD8-furin or other cargo at a level that did not (obviously) alter its normal localization, (2) contained no other obvious abnormality (and thus was deemed representative of the population) and (3) was not dividing, as evidenced by Hoechst staining. We study effects of transmembrane cargo on adaptor protein recruitment so it is important to use minimal recombinant protein expression to mimic the endogenous cargos. Thus, a fourth criterion in our studies was to choose cells with low intensity staining, compared with the general cell population. However, 3D3I is not limited to the analysis of proteins expressed at minimal levels. For example, if one wanted to evaluate organelle fragmentation in response to overexpression of a protein, this could be achieved simply by selecting cells expressing the higher levels of the protein of interest (based on intensity of staining), and compare them to cells expressing low levels or no recombinant protein. In our case we are interested in the percentage of a signal that is found within an isosurface defined by another marker but the output also gives number and volumes of isosurfaces, which may be the more relevant information to addressing different questions.
Once cells are selected, points above and below the cell in the z-plane were defined by driving the microscope to a point just out of focus on both the top and bottom of the cell. Images were recorded as a series of .tif files with dimensions of 1,316 × 1,035 pixels and a file-depth of 16 bits. Stacks of images were collected for two channels with a step-size of 0.2 μm. Exposure times of 200 ms were used for both channels. Serial images, composing the stack of images for each channel, were opened in Huygens SVI Deconvolution software, and were deconvolved using settings appropriate for Alexa594 and Alexa488 fluorophores with a total image change threshold of 0.1, block processing on, and a maximum iteration value of 40. Deconvolved images were returned in .ics format. Images were not processed in any other way prior to the performance of the calculations described.
Image analysis
Images were analyzed using Imaris6.4 software. For comparison, calculations performed using Imaris were repeated using ImageJ software where indicated. It is of note that after version 7.1, the iterative selection method of object identification is no longer the default setting. For ImageJ analysis, deconvolved .ics files were opened and converted to 16-bit files. The Co-localization Threshold plugin (http://pacific.mpi-cbg.de/wiki/index.php/Colocalization_Threshold) and the Mander’s Calculator plugin (www.macbiophotonics.ca/downloads.htm) were used to calculate Pearson’s, Mander’s M1 and M2 and tM1 and tM2 coefficients. For co-localization analysis performed using Imaris, the deconvolved .ics files were opened and the pixel dimensions were assigned to those corresponding to the wide field microscope described above (0.113 × 0.113 × 0.2 μm per voxel). The merged image was analyzed using the “Co-localization” function and the resulting co-localization coefficients were copied into a text file and saved. A mask of co-localizing pixels, and the scattergram of pixel intensities was later exported. For isosurface generation within Imaris, the image was loaded as described above. The “Generate Isosurface” function within Imaris, in which the user indicates the channel of interest, was launched and used to define the object. We generated isosurfaces using the “absolute intensity” settings without filtering. The tabular results were then exported to Excel and a “snapshot” of the scene containing the isosurface was saved. See for workflow. Statistical analyses were performed by importing the tabular results from Imaris into GraphPad Prism 5.0 and analyzed using the methods indicated.
| Abbreviations: | ||
| 3D3I | = | three-dimensional isosurface image intensity |
| MOC | = | Mander’s overlap coefficient |
| PCC | = | Pearson’s correlation coefficient |
| voxel | = | pixel with volume |
Acknowledgments
The authors would like to thank Victor Faundez for critical reading of the manuscript. This research was supported by funding from the National Institutes of General Medical Sciences (R01-GM067226) and in part by the Microscopy Core of the Emory Neuroscience NINDS Core Facilities (P30-NS055077).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Shaw PJ. Comparison of Widefield/Deconvolution and Confocal Microscopy for Three-Dimensional Imaging. In: Pawley JB, ed. Handbook of Confocal Microscopy. New York, NY: Springer Science+Business Media, 2006:453-67.
- Biggs DS. 3D deconvolution microscopy. Curr Protoc Cytom 2010; Chapter 12:Unit 12 9 1-20.
- Cannell M, McMorland A, Soeller C. Image Enhancement by Deconvolution. In: Pawley JB, ed. Handbook of Confocal Microscopy. New York, NY: Springer Science+Business Media, 2006:488-500.
- McNally JG, Karpova T, Cooper J, Conchello JA. Three-dimensional imaging by deconvolution microscopy. Methods 1999; 19:373 - 85; http://dx.doi.org/10.1006/meth.1999.0873; PMID: 10579932
- Holmes T, Biggs D, Abu-Tarif A. Blind Deconvolution. In: Pawley JB, ed. Handbook of Confocal Microscopy, 2006:468-7.
- Swedlow JR. Quantitative fluorescence microscopy and image deconvolution. Methods Cell Biol 2007; 81:447 - 65; http://dx.doi.org/10.1016/S0091-679X(06)81021-6; PMID: 17519179
- Pawley JB. Points, Pixels, and Gray Levels: Digitizing Image Data. In: Pawley JB, ed. Handbook of Confocal Microscopy. New York, NY: Springer Science+Business Media, 2006:59-79.
- Woodcroft BJ, Hammond L, Stow JL, Hamilton NA. Automated organelle-based colocalization in whole-cell imaging. Cytometry A 2009; 75:941 - 50; http://dx.doi.org/10.1002/cyto.a.20786; PMID: 19746416
- Hammond AT, Glick BS. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol Biol Cell 2000; 11:3013 - 30; PMID: 10982397
- Cayley A. On Contour and Slope Lines. Philos Mag 1859; 18:264 - 8
- Gauch JM. Image segmentation and analysis via multiscale gradient watershed hierarchies. IEEE Trans Image Process 1999; 8:69 - 79; http://dx.doi.org/10.1109/83.736688; PMID: 18262866
- Gniadek TJ, Warren G. WatershedCounting3D: a new method for segmenting and counting punctate structures from confocal image data. Traffic 2007; 8:339 - 46; http://dx.doi.org/10.1111/j.1600-0854.2007.00538.x; PMID: 17319897
- Ridler T, Calvard S. Picture thresholding using an iterative selection method. IEEE Trans Syst Man Cybern 1978; SMC-8:630 - 2
- Yang X, Beyenal H, Harkin G, Lewandowski Z. Evaluation of biofilm image thresholding methods. Water Res 2001; 35:1149 - 58; http://dx.doi.org/10.1016/S0043-1354(00)00361-4; PMID: 11268835
- Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A 2010; 77:733 - 42; http://dx.doi.org/10.1002/cyto.a.20896; PMID: 20653013
- Barlow AL, Macleod A, Noppen S, Sanderson J, Guérin CJ. Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of pearson’s correlation coefficient. Microsc Microanal 2010; 16:710 - 24; http://dx.doi.org/10.1017/S143192761009389X; PMID: 20946701
- Manders E, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual-color confocal images. J Microsc 1993; 169:375 - 82; http://dx.doi.org/10.1111/j.1365-2818.1993.tb03313.x
- Teuchert M, Schäfer W, Berghöfer S, Hoflack B, Klenk HD, Garten W. Sorting of furin at the trans-Golgi network. Interaction of the cytoplasmic tail sorting signals with AP-1 Golgi-specific assembly proteins. J Biol Chem 1999; 274:8199 - 207; http://dx.doi.org/10.1074/jbc.274.12.8199; PMID: 10075724
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 2002; 3:753 - 66; http://dx.doi.org/10.1038/nrm934; PMID: 12360192
- Shrivastava-Ranjan P, Faundez V, Fang G, Rees H, Lah JJ, Levey AI, et al. Mint3/X11gamma is an ADP-ribosylation factor-dependent adaptor that regulates the traffic of the Alzheimer’s Precursor protein from the trans-Golgi network. Mol Biol Cell 2008; 19:51 - 64; http://dx.doi.org/10.1091/mbc.E07-05-0465; PMID: 17959829
- Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol 1996; 16:6229 - 41; PMID: 8887653
- Borg JP, Yang Y, De Taddéo-Borg M, Margolis B, Turner RS. The X11alpha protein slows cellular amyloid precursor protein processing and reduces Abeta40 and Abeta42 secretion. J Biol Chem 1998; 273:14761 - 6; http://dx.doi.org/10.1074/jbc.273.24.14761; PMID: 9614075
- Andersen OM, Schmidt V, Spoelgen R, Gliemann J, Behlke J, Galatis D, et al. Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 2006; 45:2618 - 28; http://dx.doi.org/10.1021/bi052120v; PMID: 16489755
- Lodish HF, Kong N. Reversible block in intracellular transport and budding of mutant vesicular stomatitis virus glycoproteins. Virology 1983; 125:335 - 48; http://dx.doi.org/10.1016/0042-6822(83)90206-4; PMID: 6301145
- Griffiths G, Pfeiffer S, Simons K, Matlin K. Exit of newly synthesized membrane proteins from the trans cisterna of the Golgi complex to the plasma membrane. J Cell Biol 1985; 101:949 - 64; http://dx.doi.org/10.1083/jcb.101.3.949; PMID: 2863275
- Mottet G, Tuffereau C, Roux L. Reduced temperature can block different glycoproteins at different steps during transport to the plasma membrane. J Gen Virol 1986; 67:2029 - 35; http://dx.doi.org/10.1099/0022-1317-67-9-2029; PMID: 3018135
- Saraste J, Kuismanen E. Pre- and post-Golgi vacuoles operate in the transport of Semliki Forest virus membrane glycoproteins to the cell surface. Cell 1984; 38:535 - 49; http://dx.doi.org/10.1016/0092-8674(84)90508-7; PMID: 6432345
- Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol 2004; 165:111 - 22; http://dx.doi.org/10.1083/jcb.200312034; PMID: 15078902
- Valnes K, Brandtzaeg P. Retardation of immunofluorescence fading during microscopy. J Histochem Cytochem 1985; 33:755 - 61; http://dx.doi.org/10.1177/33.8.3926864; PMID: 3926864