Abstract
Keratinocyte differentiation is essential for skin development and the formation of the skin permeability barrier. This process involves an orchestrated remodelling of lipids. The cleavage of precursor lipids from lamellar bodies by β-glucocerebrosidase, sphingomyelinase, phospholipases, and sterol sulfatase generates ceramides, non-esterified fatty acids (FAs), and cholesterol for the lipid-containing extracellular matrix, the lamellar membranes in the stratum corneum. The importance of triacylglycerol (TAG) hydrolysis for the formation of a functional permeability barrier was only recently appreciated. Mice with defects in TAG synthesis (acyl-CoA:diacylglycerol acyltransferase-2-knock-out) or TAG catabolism (comparative gene identification-58, – CGI-58-knock-out) develop severe permeability barrier defects and die soon after birth because of desiccation. In humans, mutations in the CGI-58 gene also cause (non-lethal) neutral lipid storage disease with ichthyosis (NLSDI). As a result of defective TAG synthesis or catabolism, humans and mice lack ω-(O)-acylceramides, which are essential lipid precursors for the formation of the corneocyte lipid envelope. This structure plays an important role in linking the lipid-enriched lamellar membranes to highly cross-linked corneocyte proteins. This review focuses on the current knowledge of biochemical mechanisms that are essential for epidermal neutral lipid metabolism and the formation of a functional skin permeability barrier.
Introduction
Survival in a terrestrial environment necessitates the prevention of water and electrolyte loss through the body surface to minimize desiccation. In mammals, the epidermal permeability barrier in the cornified layer of the skin (stratum corneum) provides this function. The barrier is built during keratinization and consists of terminally differentiated keratinocytes (corneocytes), which are connected by protein cross-linking. These large aggregates are embedded in a lipid-containing extracellular matrix that mediates permeability barrier function.Citation1–Citation5 This prevents the excessive loss of body fluids but in addition also provides an effective barrier against environmental xenobiotics and host infections by pathogens. The most common lipids found in the extracellular matrix of the stratum corneum are ceramides, cholesterol and non-esterified fatty acids (FAs).Citation6–Citation8 They originate from enzymatic conversion of glucosylceramides, sphingomyelins, glycerophospholipids and cholesterol sulfate, which are secreted by lamellar bodies from keratinocytes at the stratum granulosum/stratum corneum interphase.Citation9–Citation13 Triacylglycerols (TAGs) are a minor lipid species of lamellar bodiesCitation10,Citation11 and as a result epidermal TAG metabolism did not obtain much attention in the past. Recent discoveries, however, demonstrated that dysfunction and deficiencies of enzymes and activators of TAG synthesis and catabolism cause major skin defects in mice and humans. These findings summarized in this review suggest a crucial role of neutral lipid metabolism in the generation of a functional permeability barrier in the skin.
Neutral Lipid Storage Disease, a Phenotypically Heterogeneous Disorder
The first indication that neutral lipid metabolism may be linked to skin function was obtained in 1966 when Rozenszajn et al.Citation14 reported on two sisters suffering from neutral lipid storage disease (NLSD) and severe ichthyosis. Later, Dorfman et al.Citation15 and Chanarin et al.Citation16 presented additional cases of NLSD patients with ichthyosis and the condition was subsequently termed Dorfman-Chanarin syndrome (DCS). NLSD is defined as a rare autosomal recessive disorder characterized by excessive deposition of neutral lipids in multiple organs of the body. The very first case report about a patient suffering from NLSD was published in the early 1950s. In this report,Citation17 Jordans documented the presence of large neutral lipid-containing vacuoles in white blood cells (“Jordans' anomaly”) of two brothers. Both individuals also suffered from a rapidly progressing skeletal and cardiac myopathy, as well as liver steatosis. In contrast to the patients reported by Dorfman et al.Citation15 and Chanarin et al.Citation16 these brothers showed no signs of ichthyosis or other skin defects suggesting genetic and clinical heterogeneity among NLSD patients. To date, a total of more than 50 NLSD cases have been reported. Besides the consistently occurring multi-systemic accumulation of TAGs in these patients, the more inconsistent clinical manifestations of the disorder include ichthyosis, skeletal and cardiac myopathy, liver steatosis with hepatosplenomegaly, neurosensory hearing loss, ataxia, horizontal nystagmus, mental retardation or developmental disorders, such as short stature, microcephalus and ear abnormalities.Citation14–Citation19
Early biochemical studies with cultured skin fibroblasts derived from DCS patients demonstrated that the total cellular TAG content was up to 20-fold higher than in controls, even when cells were cultivated in lipid-deficient media.Citation20 Biochemical experiments revealed normal FA uptake, transport and β-oxidation. With regard to TAG and phospholipid metabolism, two biochemical defects were identified: Willliams et al.Citation21, Salvayre, Douste-Blazy and colleaguesCitation22–Citation26 concluded from their studies that TAG accumulation in NLSD fibroblasts is due to a functional deficiency of a TAG lipase. In contrast, Igal and ColemanCitation27,Citation28 found that TAG hydrolysis was normal in NLSD fibroblasts but instead demonstrated that acylglycerol recycling to phospholipids was defective and caused increased DAG reesterification and TAG formation. However, the actual gene(s) and protein(s) causing NLSD remained unidentified.
Mutations in the Gene Encoding “Comparative Gene Identification-58” Cause NLSD with Ichthyosis
Important progress was made in 2001, when the group of Fischer and colleaguesCitation29 found several causative mutations in the gene coding for “comparative gene identification-58” (CGI-58) in patients with DCS. Currently, more than two dozen different mutations in the human CGI-58 gene are known including missense mutations, nonsense mutations, splice site mutations at exon-intron boundaries and insertion/deletion mutations.Citation29–Citation42 Importantly, all known NLSD patients, who are affected by mutations in the CGI-58 gene, suffer from ichthyosis. The ichthyosiform phenotype of DCS most closely resembles non-bullous congenital ichthyosiform erythroderma and is characterized by a pronounced defect in the permeability barrier function of the skin leading to an increased transepidermal water loss.Citation43,Citation44
As the name implies, CGI-58 was originally identified in a comparative-gene-identification approach, where the human proteome and the proteome of the nematode Caenorhabditis elegans (C. elegans) were aligned to detect orthologous proteins.Citation45 In the human genome, the CGI-58 gene is located on chromosome 3 and consists of 7 exons spanning a region of about 31.8 kb. Translation of the 1,050 bp mRNA results in a protein composed of 349 amino acids with a molecular mass of approximately 39 kDa. The human protein shares an amino acid homology of 94% with its murine orthologue. The highest expression of CGI-58 is observed in mature adipocytes and testes, but lower levels are also detected in liver, skeletal and cardiac muscle, skin and brain.Citation34,Citation46
Despite the causal relationship between mutations in CGI-58 and the development of DCS, the biochemical function of the protein remained elusive for a long time. CGI-58 belongs to the esterase/thioesterase/lipase subfamily of proteins, structurally defined by the presence of an α/β hydrolase fold. Accordingly, CGI-58 is also annotated as α/β hydrolase domain containing 5(ABHD5). Unlike all other members of this protein family, however, the putative active serine within the canonical esterase/lipase GXSXG sequence motif of the catalytic triad is replaced by an asparagine residue in the corresponding sequence region of CGI-58 (amino acid residue 153 of the human protein).Citation29 As a result, CGI-58 does not exhibit lipase/esterase activity in in vitro studies.Citation34,Citation47
An important function of CGI-58 was discovered in 2006. In this study Zechner and colleaguesCitation34 demonstrated that efficient hydrolysis of TAGs by adipose triglyceride lipase (ATGL) depends on the presence of CGI-58. ATGL is an essential enzyme for the hydrolytic degradation of TAGs, a process commonly named lipolysis. The complete catabolism of TAGs to FAs and glycerol requires the hydrolytic activities of three specific lipases:Citation48–Citation51 Adipose triglyceride lipase (ATGL; also termed desnutrin, patatin-like phospholipase domain containing 2,PNPLA2, or transport secretion protein-2.2,TTS-2.2) exhibits high substrate specificity for TAGs and catalyzes the initial step in lipolysis by removing the first FA from TAG molecules thereby generating diacylglycerols (DAGs). Hormone-sensitive lipase (HSL) is the major lipase in DAG hydrolysis, leading to the formation of monoacylglycerols (MAGs) and FAs. In the final step of lipolysis, monoglyceride lipase (MGL) hydrolyzes MAGs to glycerol and FAs. CGI-58 interacts with ATGL, thereby stimulating its enzymatic activity up to 20-fold.Citation34 Mutant variants of CGI-58, harboring amino acid substitutions, which lead to DCS, completely lose their ability to activate ATGL-mediated lipolysis.Citation34,Citation52 Conversely, expression of functional CGI-58 in DCS fibroblasts restored TAG hydrolysis and reversed the lipid storage phenotype in these cells.Citation34 Thus, CGI-58 appears to be an essential regulatory factor for efficient cellular TAG catabolism. Defective TAG catabolism also provided a plausible biochemical explanation for the multi-systemic TAG accumulation in patients lacking functional CGI-58.
Mutations in the ATGL Gene Cause NLSD with Myopathy but not Ichthyosis
The functional link between CGI-58 and ATGL suggested that in a subpopulation of patients NLSD may be actually caused by mutations in the ATGL gene (PNPLA2). Indeed, in 2007 Fischer et al.Citation53 published several mutations in PNPLA2 showing that ATGL dysfunction causes NLSD. To date, 14 patients with 9 different mutations have been reported in references Citation53–Citation60. All patients exhibit the classical symptoms of multi-systemic TAG accumulation. Surprisingly, however, NLSD patients with mutations in the ATGL gene do not develop ichthyosis. Instead, these patients, compared to those with mutated CGI-58, suffer from more severe forms of skeletal and cardiac myopathy. The cardiac myopathy can be lethal and requires heart transplantation in many cases.Citation58 In consideration of the phenotypical differences due to distinct genetic defects, Fischer et al.Citation53 suggested the name “NLSD with myopathy” (NLSDM) for the subgroup of patients lacking normal ATGL function and “NLSD with ichthyosis” (NLSDI) for those patients, who are affected by CGI-58 mutations (previously termed DCS).
The clinical symptoms of NLSDM resemble in many ways the phenotype of ATGL-deficient mice, i.e., excessive accumulation of TAG in multiple tissues and cell types, including heart, skeletal muscle, liver and lymphocytes.Citation61 Notably, ATGL-deficient mice just like humans suffering from NLSDM also do not display ichthyosis or other forms of skin defects. This argues against the involvement of ATGL in the development of ichthyosis and provides strong evidence for an ATGL-independent function of CGI-58 in epidermal lipid metabolism.
The ATGL-Independent Function of CGI-58 in the Skin: Role of Epidermal TAG Hydrolysis
An ATGL-independent function of CGI-58 was evident, when Radner et al.Citation62 reported on the phenotype of mice lacking CGI-58. Similar as observed in patients suffering from NLSDI, newborn CGI-58-deficient mice suffer from an ichthyosiform skin defect. In contrast to NLSDI patients, however, the defect is much more severe leading to rapid desiccation and premature death within a few hours after birth. The reason for this phenotypical difference between CGI-58-defective humans and mice is currently unknown. A plausible explanation could be that humans have a more advantageous ratio of body volume to skin surface, impeding rapid dehydration. Indirect support for this speculation came from a recently published NLSDI case report on a patient with strong seasonal variation of disease activity.Citation39 Ichthyosiform skin lesions in this subject recurred every year with beginning of summer and mostly disappeared with lower air temperatures in autumn and winter indicating more pronounced severity of disease symptoms with higher ambient temperatures.
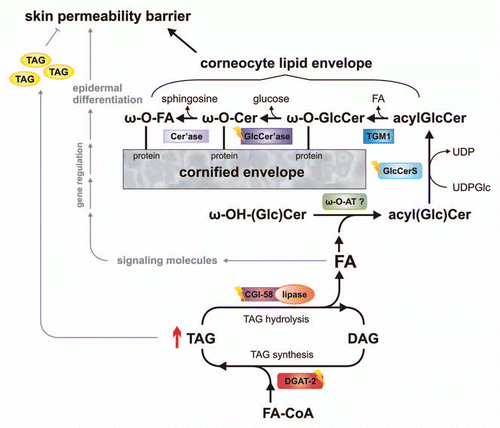
CGI-58 knockout mice also provided a suitable model to address questions regarding the biochemical defects leading to barrier dysfunction. This study demonstrated reduced TAG hydrolase activity in association with increased TAG accumulation and the absence of measurable concentrations of ω-(O)- acylceramide (acylceramides) species in the CGI-58-deficient epidermis.Citation62 The defect could be partially restored by the addition of purified CGI-58, but not by ATGL. These observations suggest that CGI-58 activates a TAG-lipase distinct from ATGL, which is required for the provision of FAs for the esterification of ω-OH-ceramides to generate acylceramides. Acylceramides are critical for the covalent binding of ω-OH-ceramides to structural proteins of corneocytes (involucrin, envoplakin and periplakin).Citation63–Citation65 This layer of protein-bound ω-OH-ceramides is referred to as corneocyte lipid envelope (CLE) and provides an interface between the cross-linked corneocyte proteins and the lipid-containing extracellular matrix. The generation of acylceramides and their involvement in the development of a functional CLE is summarized in . Acylceramides are formed from ceramides by amide bonding of very-long chain FAs to sphinganine, subsequent ω-hydroxylation of these FAs, and esterification of the ω-OH group, mostly by linoleic acid. Wertz and DowningCitation66 have demonstrated in porcine epidermis that the majority of linoleic acid for acylceramide synthesis originates from phospholipids and TAGs. In CGI-58-deficient mice this transfer apparently does not occur, leading to the accumulation of ω-OH-ceramides and the absence of acylceramides and its secondary products, like protein-bound ω-OH-ceramides ().
Importantly, these results conform to observations in the epidermis of NLSDI patients. In humans CGI-58 co-localizes in the stratum granulosum with lamellar bodies.Citation67 Lamellar body contents are secreted from keratinocytes into stratum corneum interstices and further metabolized by co-secreted enzymes, including TAG hydrolase(s),Citation68 ß-glucocerebrosidase,Citation69–Citation71 sphingomyelinase,Citation72,Citation73 and phospholipase(s).Citation74,Citation75 These enzymes are essential for the generation of the lipid constituents of the epidermal barrier.Citation76,Citation77 CGI-58 deficiency in human epidermis results in abnormal lamellar bodies in the granular layer containing neutral lipid micro-inclusions that are co-secreted with lamellar contents.Citation30,Citation78 As a consequence, the excessive incorporation of non-polar lipids into lamellar membranes forms a non-lamellar phase, which markedly impairs permeability barrier function ().Citation44 Taken together, human CGI-58-deficient epidermis lacks an essential TAG hydrolase activity to catabolize lamellar TAGs. Just as in CGI-58-deficient mice, the absence of this lipase activity causes a lack of acylceramides and protein-bound ω-OH-ceramides in the epidermis of NLSDI patientsCitation79 arguing for a defective FA transfer from TAG (or phospholipids) to ω-OH-ceramides.
Using the ORESTES (open reading frame ESTs) method, an excellent study investigating human genes involved in epidermal differentiation and barrier function, revealed a list of several potential candidates for the postulated unknown epidermal TAG lipase.Citation80 The authors identified in the human genome many potential lipase genes including members of the lipase-like family (LIPK, LIPM and LIPN), the PNPLA family (PNPLA1), the family of esterases/carboxyl esterases (CES3) and the ABHD family (ABHD9). Whether any of these genes encode lipases involved in TAG hydrolysis in a CGI-58-dependent manner remains to be explored. The classical enzymes of lipolysis (ATGL, HSL and MGL) were not detected in this study. In addition, no information on expression levels of these enzymes in the epidermis was reported. Thus, these enzymes are not expected to play a major role in epidermal TAG catabolism.
The immanent role of epidermal TAG stores in acylceramide synthesis is also evident from a mutant mouse model lacking the principal enzyme for epidermal TAG synthesis, acyl-CoA:diacylglycerol acyltransferase-2 (DGAT-2). DGAT-2-deficient mice also exhibit drastically reduced epidermal acylceramide levels and increased transepidermal water loss due to a severe permeability barrier dysfunction of the skin.Citation81 This phenotype is essentially identical to the one observed in CGI-58-deficient mice and also leads to postnatal death within a few hours after birth. In contrast to CGI-58-deficient mice, however, the skin defect in DGAT-2-deficient mice seems to include a systemic component, because transplantation of full-thickness DGAT-2-deficient skin onto the back of athymic nude mice showed a correction of the epidermal structural and functional defects.Citation81 A similar transplantation experiment with CGI-58-deficient mice failed to correct the skin phenotype arguing for a skin autonomous pathology.Citation62 Further studies are required to elucidate this interesting difference between the two mouse models. Nevertheless, the phenotypes of CGI-58- and DGAT-2-deficient mice suggest that in the epidermis FAs have to cycle through esterification (TAG synthesis) and re-hydrolysis (TAG degradation by lipases) in order to be available for ω-OH-ceramide esterification ().
The ATGL-Independent Function of CGI-58 in the Skin: CGI-58 is an Acyl-CoA:Lysophosphatidic Acid Acyltransferase
In 2008, Gosh et al.Citation82 reported that CGI-58 exhibits enzyme activity as an acyl-CoA-dependent lysophosphatidic acid acyltransferase (LPAAT) generating phosphatidic acid. This activity was later confirmed by several other groups.Citation83,Citation84 Phosphatidic acid is a key metabolite in the synthesis pathway of both TAGs and glycerophospholipidsCitation85,Citation86 and it is therefore conceivable that the CGI-58-associated LPAAT activity relates to the skin phenotype in NLSDI patients and mice with CGI-58 deficiency. Additionally, this activity coheres with previous studies by Igal and ColemanCitation27,Citation28 identifying a defect in the remodeling of TAGs to glycerophospholipids in skin fibroblasts of patients affected by NLSDI (see above). To assess whether CGI-58 affects epidermal phospholipid metabolism, Radner et al.Citation62 analyzed LPAAT activity and phospholipid composition of normal and CGI-58-deficient epidermal explants. In keratinocytes, phospholipids are usually secreted by lamellar bodies and subsequently hydrolyzed by secretory phospholipase A2 activity in the cornified layer.Citation74,Citation75 Consequently, a lack of phospholipids in the epidermis would deplete the extracellular, non-esterified FA pool and impair stratum corneum acidification.Citation87,Citation88 However, CGI-58-deficient mice exhibited normal LPAAT activity in skin lysates,Citation62 normal epidermal phospholipid composition,Citation62 and normal acidification of the stratum corneum (unpublished observation). These findings argue against a major role of CGI-58 in epidermal phospholipid metabolism.
Additionally, it is unlikely that the LPAAT activity of CGI-58 contributes to the net synthesis of TAG in keratinocytes because CGI-58-deficiency in humans and mice causes TAG accumulation.Citation39,Citation62 This is in contrast to the clinical appearance of patients lacking classical LPAAT enzymes such as acyl-CoA:acyglycerol acyltransferase (AGPAT). All of these patients suffer from the severe lack of TAGs and lipodystrophy. Moreover, mutant proteins of CGI-58 as present in patients with NLSDI retain full LPAAT activity in vitro.Citation82 However, it is conceivable that the acyltransferase activity of CGI-58 is confined to specific cellular compartments (e.g., lipid droplets or lamellar bodies) and/or substrates. For example, it may be relevant for the production of signaling lipids (e.g. phosphatidic acid) that affect keratinocyte differentiation or acylation of alternative lipid substrates, such as ω-OH-(glucosyl) ceramides, to generate acyl(glucosyl)ceramides. Although such an enzyme activity has been described in the epidermis the corresponding gene and protein remain unidentified.Citation89
The Role of CGI-58 in Keratinocyte Differentiation
Epidermal differentiation is a highly regulated, sequential process, which starts with the migration of keratinocytes from the proliferating, basal cell layer of the epidermis through the spinous and granular layer and ends with the formation of the stratum corneum.Citation90,Citation91 Using both quantitative real-time reverse transcription polymerase chain reaction and immunohistochemistry, Akiyama et al.Citation67 demonstrated a crucial role of CGI-58 in keratinocyte differentiation by showing that epidermal CGI-58 transcription is massively upregulated during cornification and fetal development of human skin. In accordance with these observations, a reduced expression of keratinization markers was observed in CGI-58 silenced keratinocytes (via siRNA) and murine CGI-58-deficient epidermis.Citation62,Citation67 The mechanisms by which CGI-58 affects these biological processes remain speculative but it is conceivable that CGI-58 is involved in the generation of lipid ligands for nuclear hormone receptors that regulate keratinocyte differentiation. Human epidermis expresses all isoforms of peroxisome proliferator-activated receptor (PPAR, isoforms: PPAR-alpha, PPAR-beta/delta, PPAR-gamma), retinoid X receptor (RXR, isoforms: RXR-alpha, RXR-beta, RXR-gamma), as well as liver X receptor (LXR, isoforms: LXR-alpha, LXR-beta).Citation92–Citation94 Activation of these receptors induces epidermal differentiation and improves permeability barrier homeostasis by stimulating epidermal lipid synthesis, lamellar body formation and secretion of lipids and enzymes, which are required for processing of extracellular lipids in the cornified layer that facilitate adequate lamellar membrane formation and consequently, normal permeability barrier function ().95
According to the current concept, PPARs and LXRs form heterodimers with RXRs. Ligand-binding causes the dissociation of co-repressors and the concurrent recruitment of co-activator proteins to the complex. This, in turn, promotes transcription of downstream target genes.Citation96,Citation97 Endogenous lipid-ligands for PPARs, LXRs, as well as RXRs include FAs, oxysterols and 9-cis retinoic acid, respectively. CGI-58 may contribute to the generation of these lipids. For example, the activation of TAG hydrolysis by CGI-58 may provide FAs for PPAR activation. The LPAAT activity of CGI-58 may lower the cellular concentration of lysophosphatidic acid and increase the levels of phosphatidic acid, thereby also affecting PPAR activity. 9-cis retinoic acid, the lipid ligand for RXR activation, derives from retinyl ester stores in keratinocytes. Its generation requires retinyl ester hydrolysis and retinol oxidation in a two-step process. Notably, retinyl ester hydrolysis in human epidermis is mediated by PNPLA4 (also annotated as gene sequence 2, GS2), a protein with high structural homology to ATGL.Citation98,Citation99
Conclusion
Recent advances in the understanding of epidermal differentiation and the formation of the skin permeability barrier acknowledge the immanent role of TAG-derived FAs in these processes. Studies with mutant mouse models and patients with NLSDI demonstrated that FAs originating from the lipolytic hydrolysis are a prerequisite for the formation of acyl(glucosyl)ceramides, which are essential for the formation of a functional CLE. However, important questions concerning the role of neutral lipid metabolism and epidermal lipid remodeling remain unanswered. For example, it is unclear why FAs must derive from TAGs for acylceramide synthesis and which CGI-58-regulated lipase mediates the hydrolysis of TAGs in keratinocytes. Future studies are required to elucidate how various activities of CGI-58 affect the formation of bioactive lipids that are involved in cellular signaling. These processes may include FA or phosphatidic acid activation of nuclear hormone receptors, which are known to control the complex and essential differentiation of keratinocytes during permeability barrier development.
Abbreviations
| ABHD 5/9 | = | α/β hydrolase domain containing protein 5/9 |
| ATGL | = | adipose triglyceride lipase |
| CES3 | = | carboxyl esterase 3 |
| CGI-58 | = | comparative gene identification-58 |
| CLE | = | corneocyte lipid envelope |
| DAG | = | diacylglycerol |
| DGAT-2 | = | acyl-CoA:diacylglycerol acyltransferase-2 |
| DCS | = | dorfman-chanarin syndrome |
| FA | = | fatty acid |
| HSL | = | hormone-sensitive lipase |
| LPAAT | = | acyl-CoA:lysophosphatidic acid acyltransferase |
| LXR | = | liver X receptor |
| MAG | = | monoacylglycerol |
| MGL | = | monoglyceride lipase |
| NLSD | = | neutral lipid storage disease |
| NLSDI | = | NLSD with ichthyosis |
| NLSDM | = | NLSD with myopathy |
| PNPLA 1/2 | = | patatin-like phospholipase domain containing 1/2 |
| PPAR | = | peroxisome proliferator-activated receptor |
| RXR | = | retinoid X receptor |
| TAG | = | triacylglycerol |
Figures and Tables
Figure 1 Schema depicting the role of epidermal triacylglycerol (TAG) metabolism in skin permeability barrier development. Acyl-CoA:diacylglycerol acyltransferase-2 (DGAT-2) transfers an activated fatty acid (FA-CoA) onto diacylglycerol (DAG) to generate TAG. These TAGs are hydrolyzed by a lipase, co-activated by comparative gene identification-58 (CGI-58). TAG-derived FAs are then activated and transferred by an unknown ω-(O)-acyltransferase (ω-O-AT?) onto ω-OH-(glucosyl)ceramides (ω-OH-(Glc)Cer). Acylceramides (acylCer) are glucosylated by uridine diphosphate glucose:ceramide glucosyltransferase (glucosylceramide synthase, GlcCerS). Generated acylglucosylceramides (acylGlcCer) are then covalently bound to proteins of the cornified envelope by a transesterification reaction (transglutaminase 1, TGM1), deglucosylated (β-glucocerebrosidase-GlcCer'ase) and hydrolyzed (ceramidase, Cer'ase). TAG-derived FAs may also act as ligands for nuclear hormone receptors, activating transcriptional regulators involved in epidermal differentiation. Excessive epidermal TAG accumulation (as indicated by ↑) may lead to lamellar/nonlamellar phase separation and permeability barrier dysfunction. Defects in protein function (as indicated by flashes) result in ichthyosiform phenotypes.
Acknowledgements
This work was supported by the grant “GOLD-Genomics of Lipid-Associated Disorders,” which is part of the Austrian Genome Project “GEN-AU Genome research in Austria,” funded by the Austrian Ministry for Science and Research and the FFG. This research was also supported by the grants SFB LIPOTOX (F30) and the Wittgenstein Award Z136, which are funded by the Austrian Science Foundation. Additional funding was obtained from the City of Graz, the Province of Styria, and the European commission grant agreements no. 201608 (TOBI) and 202272 (LipidomicNet). We thank Mag. Dr. Gabriele Schoiswohl and Mag. Caroline Schober-Trummler for careful and critical reading of the manuscript.
References
- Nemes Z, Steinert PM. Bricks and mortar of the epidermal barri. Exp Mol Med 1999; 31:5 - 19
- Kalinin AE, Kajava AV, Steinert PM. Epithelial barrier function: assembly and structural features of the cornified cell envelope. Bioessays 2002; 24:789 - 800
- Madison KC. Barrier function of the skin: “la raison d'etre” of the epidermis. J Invest Dermatol 2003; 121:231 - 241
- Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol 2008; 17:1063 - 1072
- Jensen JM, Proksch E. The skin's barrier. G Ital Dermatol Venereol 2009; 144:689 - 700
- Wertz PW. Epidermal lipids. Semin Dermatol 1992; 11:106 - 113
- Wertz PW. Elias PM, Feingold KR. Biochemistry of human stratum corneum lipids. Skin Barrier 2006; New York, NY Taylor & Francis Group 33 - 42
- Feingold KR. The outer frontier: the importance of lipid metabolism in the skin. J Lipid Res 2009; 50:417 - 422
- Wertz PW, Downing DT, Freinkel RK, Traczyk TN. Sphingolipids of the stratum corneum and lamellar granules of fetal rat epidermis. J Invest Dermatol 1984; 83:193 - 195
- Freinkel RK, Traczyk TN. Lipid composition and acid hydrolase content of lamellar granules of fetal rat epidermis. J Invest Dermatol 1985; 85:295 - 298
- Grayson S, Johnson-Winegar AG, Wintroub BU, Isseroff RR, Epstein E Jr, Elias PM. Lamellar bodyenriched fractions from neonatal mice: preparative techniques and partial characterization. J Invest Dermatol 1985; 85:289 - 294
- Schmitz G, Muller G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J Lipid Res 1991; 32:1539 - 1570
- Elias PM, Feingold KR, Fartasch M. Elias PM, Feingold KR. Epidermal lamellar body as a multifunctional secretory organelle. Skin Barrier 2006; New York, NY Taylor & Francis Group 261 - 272
- Rozenszajn L, Klajman A, Yaffe D, Efrati P. Jordans' anomaly in white blood cells. Report of case. Blood 1966; 28:258 - 265
- Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 1974; 110:261 - 266
- Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J 1975; 1:553 - 555
- Jordans GH. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB). Acta Med Scand 1953; 145:419 - 423
- Igal RA, Rhoads JM, Coleman RA. Neutral lipid storage disease with fatty liver and cholestasis. J Pediatr Gastroenterol Nutr 1997; 25:541 - 547
- Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab 2009; 297:289 - 296
- Williams ML, Monger DJ, Rutherford SL, Hincenbergs M, Rehfeld SJ, Grunfeld C. Neutral lipid storage disease with ichthyosis: lipid content and metabolism of fibroblasts. J Inherit Metab Dis 1988; 11:131 - 143
- Williams ML, Coleman RA, Placezk D, Grunfeld C. Neutral lipid storage disease: a possible functional defect in phospholipid-linked triacylglycerol metabolism. Biochim Biophys Acta 1991; 1096:162 - 169
- Salvayre R, Negre A, Radom J, Douste-Blazy L. Independence of triacylglycerol-containing compartments in cultured fibroblasts from Wolman disease and multisystemic lipid storage myopathy. FEBS Lett 1989; 250:35 - 39
- Radom J, Salvayre R, Negre A, Maret A, Douste-Blazy L. Metabolism of neutral lipids in cultured fibroblasts from multisystemic (or type 3) lipid storage myopathy. Eur J Biochem 1987; 164:703 - 708
- Radom J, Salvayre R, Maret A, Negre A, Douste-Blazy L. Metabolism of 1-pyrenedecanoic acid and accumulation of neutral fluorescent lipids in cultured fibroblasts of multisystemic lipid storage myopathy. Biochim Biophys Acta 1987; 920:131 - 139
- Radom J, Salvayre R, Levade T, Douste-Blazy L. Influence of chain length of pyrene fatty acids on their uptake and metabolism by Epstein-Barr-virustransformed lymphoid cell lines from a patient with multisystemic lipid storage myopathy and from control subjects. Biochem J 1990; 269:107 - 113
- Hilaire N, Salvayre R, Thiers JC, Bonnafe MJ, Negre-Salvayre A. The turnover of cytoplasmic triacylglycerols in human fibroblasts involves two separate acyl chain length-dependent degradation pathways. J Biol Chem 1995; 270:27027 - 27034
- Igal RA, Coleman RA. Acylglycerol recycling from triacylglycerol to phospholipid, not lipase activity, is defective in neutral lipid storage disease fibroblasts. J Biol Chem 1996; 271:16644 - 16651
- Igal RA, Coleman RA. Neutral lipid storage disease: a genetic disorder with abnormalities in the regulation of phospholipid metabolism. J Lipid Res 1998; 39:31 - 43
- Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet 2001; 69:1002 - 1012
- Akiyama M, Sawamura D, Nomura Y, Sugawara M, Shimizu H. Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin syndrome. J Invest Dermatol 2003; 121:1029 - 1034
- Srinivasan R, Hadzic N, Fischer J, Knisely AS. Steatohepatitis and unsuspected micronodular cirrhosis in Dorfman-Chanarin syndrome with documented ABHD5 mutation. J Pediatr 2004; 144:662 - 665
- Pujol RM, Gilaberte M, Toll A, Florensa L, Lloreta J, Gonzalez-Ensenat MA, et al. Erythrokeratoderma variabilis-like ichthyosis in Chanarin-Dorfman syndrome. Br J Dermatol 2005; 153:838 - 841
- Schleinitz N, Fischer J, Sanchez A, Veit V, Harle JR, Pelissier JF. Two new mutations of the ABHD5 gene in a new adult case of Chanarin Dorfman syndrome: an uncommon lipid storage disease. Arch Dermatol 2005; 141:798 - 800
- Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 2006; 3:309 - 319
- Ben Selma Z, Yilmaz S, Schischmanoff PO, Blom A, Ozogul C, Laroche L, et al. A novel S115G mutation of CGI-58 in a Turkish patient with Dorfman-Chanarin syndrome. J Invest Dermatol 2007; 127:2273 - 2276
- Bruno C, Bertini E, Di Rocco M, Cassandrini D, Ruffa G, De Toni T, et al. Clinical and genetic characterization of Chanarin-Dorfman syndrome. Biochem Biophys Res Commun 2008; 369:1125 - 1128
- Badeloe S, van Geel M, Nagtzaam I, Rubio-Gozalbo ME, Oei RL, Steijlen PM, et al. Chanarin-Dorfman syndrome caused by a novel splice site mutation in ABHD5. Br J Dermatol 2008; 158:1378 - 1380
- Ronchetti A, Prati D, Pezzotta MG, Tavian D, Colombo R, Callea F, et al. Severe steatohepatitis in a patient with a rare neutral lipid storage disorder due to ABHD5 mutation. J Hepatol 2008; 49:474 - 477
- Ujihara M, Nakajima K, Yamamoto M, Teraishi M, Uchida Y, Akiyama M, et al. Epidermal triglyceride levels are correlated with severity of ichthyosis in Dorfman-Chanarin syndrome. J Dermatol Sci 2010; 57:102 - 107
- Emre S, Unver N, Evans SE, Yuzbasioglu A, Gurakan F, Gumruk F, et al. Molecular analysis of Chanarin-Dorfman syndrome (CDS) patients: Identification of novel mutations in the ABHD5 gene. Eur J Med Genet 2010; 53:141 - 144
- Cakir M, Bruno C, Cansu A, Cobanoglu U, Erduran E. Liver cirrhosis in an infant with Chanarin-Dorfman syndrome caused by a novel splice-site mutation in ABHD5. Acta Paediatr 2010; 99:1592 - 1594
- Redaelli C, Coleman RA, Moro L, Sertedaki A, Kakourou T, Elsayed SM, et al. Clinical and genetic characterization of Chanarin-Dorfman Syndrome patients: first report of large deletions in the ABHD5 gene. Orphanet J Rare Dis 2010; 5:33
- Williams ML. Ichthyosis: mechanisms of disease. Pediatr Dermatol 1992; 9:365 - 368
- Demerjian M, Crumrine DA, Milstone LM, Williams ML, Elias PM. Barrier dysfunction and pathogenesis of neutral lipid storage disease with ichthyosis (Chanarin-Dorfman syndrome). J Invest Dermatol 2006; 126:2032 - 2038
- Lai CH, Chou CY, Ch'ang LY, Liu CS, Lin W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res 2000; 10:703 - 713
- Subramanian V, Rothenberg A, Gomez C, Cohen AW, Garcia A, Bhattacharyya S, et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem 2004; 279:42062 - 42071
- Yamaguchi T, Omatsu N, Morimoto E, Nakashima H, Ueno K, Tanaka T, et al. CGI-58 facilitates lipolysis on lipid droplets but is not involved in the vesiculation of lipid droplets caused by hormonal stimulation. J Lipid Res 2007; 48:1078 - 1089
- Watt MJ, Steinberg GR. Regulation and function of triacylglycerol lipases in cellular metabolism. Biochem J 2008; 414:313 - 325
- Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res 2009; 50:3 - 21
- Zimmermann R, Lass A, Haemmerle G, Zechner R. Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim Biophys Acta 2009; 1791:494 - 500
- Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 2011; 50:14 - 27
- Yamaguchi T, Omatsu N, Matsushita S, Osumi T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem 2004; 279:30490 - 30497
- Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet 2007; 39:28 - 30
- Akiyama M, Sakai K, Ogawa M, McMillan JR, Sawamura D, Shimizu H. Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve 2007; 36:856 - 859
- Kobayashi K, Inoguchi T, Maeda Y, Nakashima N, Kuwano A, Eto E, et al. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J Clin Endocrinol Metab 2008; 93:2877 - 2884
- Ohkuma A, Nonaka I, Malicdan MC, Noguchi S, Ohji S, Nomura K, et al. Distal lipid storage myopathy due to PNPLA2 mutation. Neuromuscul Disord 2008; 18:671 - 674
- Campagna F, Nanni L, Quagliarini F, Pennisi E, Michailidis C, Pierelli F, et al. Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem Biophys Res Commun 2008; 377:843 - 846
- Hirano K, Ikeda Y, Zaima N, Sakata Y, Matsumiya G. Triglyceride deposit cardiomyovasculopathy. N Engl J Med 2008; 359:2396 - 2398
- Akman HO, Davidzon G, Tanji K, Macdermott EJ, Larsen L, Davidson MM, et al. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul Disord 2010; 20:397 - 402
- Chen J, Hong D, Wang Z, Yuan Y. A novel PNPLA2 mutation causes neutral lipid storage disease with myopathy (NLSDM) presenting muscular dystrophic features with lipid storage and rimmed vacuoles. Clin Neuropathol 2010; 29:351 - 356
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006; 312:734 - 737
- Radner FP, Streith IE, Schoiswohl G, Schweiger M, Kumari M, Eichmann TO, et al. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J Biol Chem 2010; 285:7300 - 7311
- Behne M, Uchida Y, Seki T, de Montellano PO, Elias PM, Holleran WM. Omega-hydroxyceramides are required for corneocyte lipid envelope (CLE) formation and normal epidermal permeability barrier function. J Invest Dermatol 2000; 114:185 - 192
- Stewart ME, Downing DT. The omega-hydroxyceramides of pig epidermis are attached to corneocytes solely through omega-hydroxyl groups. J Lipid Res 2001; 42:1105 - 1110
- Uchida Y, Holleran WM. Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci 2008; 51:77 - 87
- Wertz PW, Downing DT. Metabolism of linoleic acid in porcine epidermis. J Lipid Res 1990; 31:1839 - 1844
- Akiyama M, Sakai K, Takayama C, Yanagi T, Yamanaka Y, McMillan JR, et al. CGI-58 is an alpha/betahydrolase within lipid transporting lamellar granules of differentiated keratinocytes. Am J Pathol 2008; 173:1349 - 1360
- Menon GK, Grayson S, Elias PM. Cytochemical and biochemical localization of lipase and sphingomyelinase activity in mammalian epidermis. J Invest Dermatol 1986; 86:591 - 597
- Holleran WM, Takagi Y, Menon GK, Legler G, Feingold KR, Elias PM. Processing of epidermal glucosylceramides is required for optimal mammalian cutaneous permeability barrier function. J Clin Invest 1993; 91:1656 - 1664
- Holleran WM, Ginns EI, Menon GK, Grundmann JU, Fartasch M, McKinney CE, et al. Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease. J Clin Invest 1994; 93:1756 - 1764
- Holleran WM, Takagi Y, Menon GK, Jackson SM, Lee JM, Feingold KR, et al. Permeability barrier requirements regulate epidermal beta-glucocerebrosidase. J Lipid Res 1994; 35:905 - 912
- Jensen JM, Schutze S, Forl M, Kronke M, Proksch E. Roles for tumor necrosis factor receptor p55 and sphingomyelinase in repairing the cutaneous permeability barrier. J Clin Invest 1999; 104:1761 - 1770
- Schmuth M, Man MQ, Weber F, Gao W, Feingold KR, Fritsch P, et al. Permeability barrier disorder in Niemann-Pick disease: sphingomyelin-ceramide processing required for normal barrier homeostasis. J Invest Dermatol 2000; 115:459 - 466
- Mao-Qiang M, Feingold KR, Jain M, Elias PM. Extracellular processing of phospholipids is required for permeability barrier homeostasis. J Lipid Res 1995; 36:1925 - 1935
- Mao-Qiang M, Jain M, Feingold KR, Elias PM. Secretory phospholipase A2 activity is required for permeability barrier homeostasis. J Invest Dermatol 1996; 106:57 - 63
- Wertz PW. Lipids and barrier function of the skin. Acta Derm Venereol Suppl 2000; 208:7 - 11
- Feingold KR. Thematic review series: skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis. J Lipid Res 2007; 48:2531 - 2546
- Elias PM, Williams ML. Neutral lipid storage disease with ichthyosis. Defective lamellar body contents and intracellular dispersion. Arch Dermatol 1985; 121:1000 - 1008
- Uchida Y, Cho Y, Moradian S, Kim J, Nakajima K, Crumrine D, et al. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J Invest Dermatol 2010; 130:2497 - 2499
- Toulza E, Mattiuzzo NR, Galliano MF, Jonca N, Dossat C, Jacob D, et al. Large-scale identification of human genes implicated in epidermal barrier function. Genome Biol 2007; 8:107
- Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM, et al. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J Biol Chem 2004; 279:11767 - 11776
- Ghosh AK, Ramakrishnan G, Chandramohan C, Rajasekharan R. CGI-58, the causative gene for Chanarin-Dorfman syndrome, mediates acylation of lysophosphatidic acid. J Biol Chem 2008; 283:24525 - 24533
- Montero-Moran G, Caviglia JM, McMahon D, Rothenberg A, Subramanian V, Xu Z, et al. CGI-58/ABHD5 is a coenzyme A-dependent lysophosphatidic acid acyltransferase. J Lipid Res 2010; Apr;51:709 - 719
- Gruber A, Cornaciu I, Lass A, Schweiger M, Poeschl M, Eder C, et al. The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J Biol Chem 2010; 285:12289 - 12298
- Peterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet 2001; 27:121 - 124
- Han GS, Wu WI, Carman GM. The Saccharomyces cerevisiae Lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J Biol Chem 2006; 281:9210 - 9218
- Mauro T, Holleran WM, Grayson S, Gao WN, Man MQ, Kriehuber E, et al. Barrier recovery is impeded at neutral pH, independent of ionic effects: implications for extracellular lipid processing. Arch Dermatol Res 1998; 290:215 - 222
- Fluhr JW, Kao J, Jain M, Ahn SK, Feingold KR, Elias PM. Generation of free fatty acids from phospholipids regulates stratum corneum acidification and integrity. J Invest Dermatol 2001; 117:44 - 51
- Takagi Y, Nakagawa H, Matsuo N, Nomura T, Takizawa M, Imokawa G. Biosynthesis of acylceramide in murine epidermis: characterization by inhibition of glucosylation and deglucosylation and by substrate specificity. J Invest Dermatol 2004; 122:722 - 729
- Reichert U, Michel S, Schmidt R. Darmon M, Blumenberg M. The cornified envelope: a key structure of terminally differentiating keratinocytes. Molecular Biology of the Skin: the Keratinocyte 1993; San Diego, CA Academic Press, Inc. 107 - 150
- Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005; 6:328 - 340
- Reichrath J, Mittmann M, Kamradt J, Muller SM. Expression of retinoid-X receptors (-alpha, -beta, -gamma) and retinoic acid receptors (-alpha, -beta, -gamma) in normal human skin: an immunohistological evaluation. Histochem J 1997; 29:127 - 133
- Rivier M, Safonova I, Lebrun P, Griffiths CE, Ailhaud G, Michel S. Differential expression of peroxisome proliferator-activated receptor subtypes during the differentiation of human keratinocytes. J Invest Dermatol 1998; 111:1116 - 1121
- Russell LE, Harrison WJ, Bahta AW, Zouboulis CC, Burrin JM, Philpott MP. Characterization of liver X receptor expression and function in human skin and the pilosebaceous unit. Exp Dermatol 2007; 16:844 - 852
- Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic review series: skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res 2008; 49:499 - 509
- Hanley K, Jiang Y, He SS, Friedman M, Elias PM, Bikle DD, et al. Keratinocyte differentiation is stimulated by activators of the nuclear hormone receptor PPARalpha. J Invest Dermatol 1998; 110:368 - 375
- Hanley K, Ng DC, He SS, Lau P, Min K, Elias PM, et al. Oxysterols induce differentiation in human keratinocytes and increase Ap-1-dependent involucrin transcription. J Invest Dermatol 2000; 114:545 - 553
- Gao J, Simon M. Identification of a novel keratinocyte retinyl ester hydrolase as a transacylase and lipase. J Invest Dermatol 2005; 124:1259 - 1266
- Gao JG, Simon M. A comparative study of human GS2, its paralogues and its rat orthologue. Biochem Biophys Res Commun 2007; 360:501 - 506