Abstract
MeDIP (Methylated DNA Immunoprecipitation) is a relatively recent technique aimed to enrich the methylated fraction of DNA with an antibody directed against 5-methyl-cytosine. MeDIP processed samples are suitable for investigation of the methylation status of specific genomic loci and for performing genome-wide screening when hybridized to DNA methylation microarrays or analyzed by deep sequencing. Here, we describe a standardization protocol and quality controls to assess the specificity, reproducibility and efficiency of the MeDIP procedure. These may have utility when comparing results between samples and experiments within laboratories and between laboratories.
Introduction
DNA methylation is a widespread DNA modification in which a cytosine residue in DNA is converted enzymatically to a 5-methyl-cytosine (5-mC). In mammals, it mainly occurs in places where the cytosine residue is followed by a guanine residue (CpG). Although the CpG dinucleotide is under-represented in mammalian genomes overall, unusually dense clusters of CpG dinucleotides, which are called “CpG islands,” occur in specific genomic regions, notably in the promoter regions of genes. CpG islands are defined as sequences longer than 500 bp with CG content of at least 55% and a ratio observed/expected of CpG frequency of at least 0.6.Citation1 Using these criteria, CpG islands overlap promoter regions of 60–70% of human genesCitation2,Citation3 and methylation of these islands plays an important role in the regulation of gene expression.Citation3-Citation5 DNA methylation is a critical determinant of many biological and cellular processes, including embryonic development,Citation6 regulation of transcription,Citation7 X chromosome inactivation,Citation8 genomic imprinting and dosage compensationCitation9 and genome defense from molecular parasites.Citation10
Altered patterns of DNA methylation are associated with altered patterns of gene transcription, which are associated with, and may be causal for some, diseases.Citation11-Citation14 In addition, there is growing evidence that DNA methylation patterns change during aging.Citation15-Citation19 Therefore, the study of these changes is becoming widespread and a number of laboratory techniques have been developed to investigate specific DNA regions (or genes) and to carry out genome-wide analysis.
Many techniques are now available for characterizing the extent of DNA methylation.Citation20 The total methyl-cytosine content of DNA can be quantified by HPLCCitation21 and indices of the degree of methylation can be obtained using methyl-sensitive restriction enzymes as in the LUminometric Methylation Assay technique (LUMA).Citation22,Citation23 Other approaches take advantage of the selective deamination of unmethylated cytosine to uracil by sodium bisulfite under alkaline conditions.Citation24 Examples of these techniques are the COBRA assay,Citation25 methyl-specific PCR,Citation26 MethyLightCitation27 and pyrosequencing.Citation28
More recently, the development of the so-called ChIP-on-chip approach relies on affinity purification of the methylated fraction of the genome with an antibody or a methyl-binding protein that recognizes 5-methyl-cytidine. In the Methylated DNA Immunoprecipitation (MeDIP) method,Citation5,Citation29 total genomic DNA is sheared into random fragments, which are then immunoprecipitated with an antibody that recognizes 5-methyl-cytidine. The starting sample, called Input, contains both methylated and unmethylated fragments, while the MeDIP fraction contains mainly methylated fragments. After the immunoprecipitation reaction, the Input and the MeDIP fraction can be labeled with different dyes and co-hybridized as a two-color experiment to microarrays of (usually) CpG-rich genomic regions to obtain an estimate of genome-wide methylation pattern.
Alternatively, samples can be analyzed by semi-quantitative PCR or by real time qPCR to calculate the relative enrichment of a specific sequence between the MeDIP fraction and the whole sample (Input).
In such experiments, it is usual to assess the efficacy and repeatability of the MeDIP step by enrichment estimation for genomic regions expected to be heavily methylated in the samples under investigation. This is done from qPCR data, comparing the pairs MeDIP/Input and normalizing with genomic regions expected to be unmethylated. However, the methylation status of specific genomic regions is not always known for particular samples under the conditions of the specific study, which may make the choice of genomic regions for quality control problematical. To circumvent these problems, we have developed a set of control DNA samples [a positive (highly methylated) and a negative (unmethylated)] to be spiked into each immunoprecipitation reaction. Estimation of the relative enrichments (using qPCR) of these positive and negative controls in MeDIP/Input pairs from qPCR data allows researchers to verify that each batch of samples was enriched for methylated regions successfully and to ensure experimental reproducibility between samples and experiments within laboratories and between laboratories.
Results and Discussion
In the present study, we developed standardization protocols and quality controls to assess the specificity, reproducibility and efficiency of the MeDIP procedure. Our aim was to develop and test protocols and quality controls that may have utility when comparing results between samples and experiments within laboratories and between laboratories. summarizes the major steps in the MeDIP procedure, which were investigated in the present study.
Figure 1. Methylated DNA Immunoprecipitation (MeDIP) protocol. The black arrow indicates the introduction of exogenous controls described in this study.
Genomic DNA extraction and Sonication
Genomic DNA was run on agarose gels to confirm (1) the integrity of each DNA sample and (2) the absence of RNA. The latter is critical to avoid antibody capture by 5-mC that is known to be present in this nucleic acid.Citation30-Citation33 Sonication patterns were assessed on agarose gels and optimally fragmented DNA samples, i.e., with fragment sizes 200 to 1000 bp,Citation29,Citation5 were purified using silica columns. We propose this method of purification because it is faster than the ethanol-based precipitation method proposed in the original method.Citation29
Preparation of methylated and unmethylated controls and assessment of utility
Methylated and unmethylated controls were prepared by PCR from unmethylated and commercially available Lambda DNA, as reported in a previous study.Citation34 We designed primers (Primer Pair 1, see ) to amplify two fragments of similar length and content of CpG (Fragments A and B). The length chosen was the average size of the sheared fragments to be used in the immunoprecipitation process (about 600 bp) and the CpG dinucleotides count was 41 for Fragment A and 35 for Fragment B. For the preparation of the positive control, Fragment A was In Vitro Methylated (IVM) using E. coli M.SssI methyltransferase, in the presence of S-Adenosyl Methionine (SAM). Fragment B was used as the negative control without any modification. The efficiency of Fragment A IVM was checked for 14 CpG sites by two methylation-sensitive restriction enzymes: AciI, which cuts blunt at CC↓CG sites (9 in the sequence) and HpaII, which cuts blunt at CC↓GG sites (5 in the sequence). Both enzymes cut DNA at the respective sequences only if the internal C is unmethylated. As a double check, we also used a third enzyme, the HpaII-isoschizomer MspI, whose cutting is not affected by the internal C methylation. We ran the same set of digestions with Fragment B, which contains 6 AciI sites and 2 MspI/HpaII sites. As expected, Fragment A IVM was cut by MspI (, lane 2), but uncut by AciI (, lane 1) or by HpaII (, lane 3): the gel shows a single band of 625 bp long in these last two cases, as for the undigested fragment (, lane 4). Full length Fragment B (, lane 8), instead, was fully digested by AciI, MspI and HpaII (lanes 5, 6 and 7, respectively).
Table 1. Primers sequences for Spike Positive and Negative Controls
Figure 2. Digestion patterns of positive and negative controls with the restriction enzymes AciI, MspI and HpaII on 2% Metaphor agarose gel. Lanes 1 and 5 = AciI digestions of positive and negative control, respectively; lane 2 and 6 = MspI digestions of positive and negative control; lanes 3 and 7 = HpaII digestions of positive and negative control; lanes 4 and 8 = undigested positive and negative control; lane 9 = 100 bp DNA Ladder. Image in the bottom row shows virtual gel with the corresponding digestions patterns.
Development and validation of primer pairs for quantification of enrichment of methylated DNA following MeDIP
For the quantification of enrichment of methylated DNA derived from the MeDIP procedure with data from qPCR, it was necessary to demonstrate that the primer pairs chosen to amplify both the methylated and unmethylated control DNA fragments (see , primer pairs 2 for sequences) amplified the selected regions linearly and also showed good efficiencies. As shown in Figure S1 A and B, the amplification linearity ranged over at least eight orders of magnitude for both controls (500 pg/μL to 50 ag/μL) and the Cq values obtained were in an acceptable scale (5 < Cq < 40). In addition, the amplification efficiencies were good for both positive control (0.82) and negative control (0.85). We concluded that the chosen primer pairs were suitable for quantification of both methylated and unmethylated DNA controls and for estimation of enrichment using the Pfaffl method.Citation35 Also, as the two efficiencies were similar, the user could simplify the enrichment calculation using the ΔΔCq approximation method.
In addition, we found that the chosen primer pairs did not result in amplification of any unspecific DNA sequences or in false priming of human and mouse gDNA at the template dilutions used (data not shown).
The next step was to determine the optimal sample concentration to be used in the qPCR assays for quantification of the controls and to ensure that amplification efficiencies for the controls was similar when tested in the context of genomic DNA to be immunoprecipitated (DNA from mouse liver) as when tested alone. We checked the amplification efficiencies of the controls in three 10-fold dilutions (100 pg/μL, 10 pg/μL and 1 pg/μL) of both Input and MeDIP samples from one of our immunoprecipitation experiments. As shown in Figure S1 C and D, we observed very similar efficiencies in the presence of both Input DNA and of methylation enriched (MeDIP) DNA for both positive and negative controls. We conclude that a) the presence of Input and MeDIP DNA fractions does not influence the amplification efficiencies of the chosen primer sets for the controls and b) 20 pg of template DNA is an acceptable amount for control PCR reactions to ensure that the answers will be in the linear range of the assay.
Methylated DNA Immunoprecipitation (MeDIP)
As described in the Materials and Methods section, in preparation for MeDIP, we spiked genomic DNA with both positive and negative control fragments in the ratio 1:100 controls:genomic DNA. This resulted in Cq values ranging from about 14 to 23 for all our subsequent qPCR assays (data not shown). From these data, we infer that users of our approach could reduce the proportions of control “spikes” to as low as 1:10000 in proportion to the total amount of sample DNA being investigated. However, the spiked amount that we suggest is high enough to allow detection using a minimal amount of IP reactions and is low enough to result in an average total controls occupancy of 5.07% for pre-Whole Genome Amplification (WGA) samples and 3.32% for post-WGA samples in the IPed samples (calculated from qPCR data, not shown). Since lambda DNA sequences do not share homology with human or mouse sequences,Citation34 they will not hybridize to human or mouse array chips and so do not interfere with downstream analysis using an array approach. However, if deep sequencing is intended to be used for further interrogation of MeDIP samples, we suggest further optimization of the fragment lengths and amounts of spikes used to limit any potential redundancy in material to be sequenced.Citation34,Citation36,Citation37
Whole Genome Amplification (WGA) and positive control/negative control enrichments
To reduce experimental costs in terms of reagents and starting material (test sample DNA), Whole Genome Amplification (WGA)Citation38 is a very useful (if not essential) step in the preparation of DNA for methylation analysis following MeDIP. The recovery of DNA following immunoprecipitation equates to only 3% to 5% of the starting material (data not shown), which is not enough for qPCR assays on endogenous regions of interest or for hybridization to microarrays so that WGA is performed routinely on DNA obtained after MeDIP. However, since our spiked controls were detectable at a very low concentration, we could measure enrichments of the positive control by comparison with the negative control after MeDIP in both pre-WGA samples and in post-WGA samples.
As shown in , we observed that enrichment of the positive control (compared with the negative control) was consistently lower after WGA and, on average, we saw a “loss” of about 32% in enrichment after WGA. This “loss” was manifest as an increase in the Cq values for the positive control of about 5 cycles, when the same sample (Input or MeDIP) was tested pre-WGA and post-WGA, while Cq values for the negative control remained similar pre- and post-WGA (data not shown). This loss is due to sequence related bias in the PCR amplification step of the WGA procedure: indeed, the ratio of Fragment A:Fragment B after WGA was always about one sixth of the ratio before WGA, irrespective of the methylation status of the fragments (data not shown). We hypothesize that the annealing/extension temperature of the amplification step in the WGA procedure might not provide enough energy to unfold stable hairpins and helixes in the context of Fragment A. To further address whether the bias introduced by WGA amplification was related to fragment length, we spiked 3 randomly chosen sonicated gDNAs with six lambda unmethylated DNA fragments of defined lengths (in the range suggested by the WGA kit manufacturer, i.e., 100–1000 bp) and measured the amplification rate of those post-WGA/pre-WGA. As shown in , there was no relationship between fragment length and amplification rate, thus confirming that the major biases during the WGA process are related to sequence rather than to fragment length. Primers sequences are listed in Table S1.
Figure 3. Panel A and C: descriptive statistics and regression analysis for fold enrichments positive control/negative control pre-WGA and post-WGA. Box plots in Panel A show the percentiles and the median of column data. Box outlines define the 25th and 75th percentiles, with a line at the median and whiskers indicate the 10th and 90th percentiles. Panel C shows linear regression for pairs of pre-WGA and post-WGA enrichments. n = 33. Panel B: WGA amplification bias. Black bars represent the amplification rate post-WGA/pre-WGA of different length Lambda DNA fragments spiked in mouse genomic DNA. Error bars show SEM, n = 3.
Although having demonstrated the presence of bias in the procedure, pre-WGA and post-WGA data distributions were very similar () and there was a strong, and significant, linear relationship between the enrichment of positive control DNA by MeDIP before and after WGA (ANOVA, p < 0.01, ). Also, in accordance to the manufacturer’s guidelines, yields of DNA following WGA were in the range of 300–700 fold of the template DNA (data not shown). Therefore, we conclude that the WGA step is a valid, and economical, method for the preparation of large amounts of MeDIP samples to be used for qualitative studies, but it is not likely to be optimal for quantitative studies. If samples amplified with such procedures are intended for quantitative studies, we recommend that preliminary experiments be performed to determine the amount of enrichment “lost” during WGA.
Enrichment of endogenous DNA sequences following MeDIP
Having demonstrated that the positive control “spike” is enriched substantially by MeDIP when compared with the negative control and that this enrichment is maintained at a constant ratio following WGA, the next step was to examine the effectiveness of the MeDIP procedure in enriching known endogenous methylated regions of the mammalian genomeCitation39-Citation42 in comparison with those endogenous regions in which little or no methylation was anticipated (normalizers).Citation29 For the latter, we chose the promoters of housekeeping genes. This is a commonly used approach, but probably not the best one, as demonstrated in this study. Primers sequences are listed in Table S2.
We tested the reproducibility of our assays and investigated the extent of the technical error on a subset of technical replicates (i.e., MeDIPs performed on human genomic DNA from MRC5 cells), by comparing estimates of GAPDH-normalized enrichments of H19 and HIST1H2BA genes () with those obtained for enrichment of the positive control with reference to the negative control for post-WGA samples (). Percentage coefficient of variations for these assays ranged from 6% to 15%, indicating that the technical error of assays of the spike controls is similar to that for endogenous regions.
Figure 4. Gene specific enrichments after MeDIP followed by WGA in MRC5 fibroblast DNA and in various mouse tissues. (A) Enrichment of the MRC5 internal genes H19 (black bar) and HIST1H2BA (gray bar), both normalized to GAPDH. (B) positive/negative control Enrichment Post-WGA. Error bars show standard error of the means, n = 3 technical replicates. Gene specific enrichments for H19 (C) and Hist1h2ba (D) in mice tissues normalized to either Actb (black bars) or Gapdh (gray bars). Data are mean ± SD; n, 3 biological replicates; AL, ad libitum fed; DR, dietary restricted.
Although we chose as normalizers housekeeping genes for which we anticipated little or no methylation in promoter or intragenic regions, it is possible that some of these normalizers may be methylated to some extent in specific tissues, or animals, under specific circumstances. To examine this possibility, we investigated mouse genomic DNA obtained from 3 different tissues (heart, liver and colon), of animals of different ages and exposed to different dietary regimens. For our calculations of enrichment of endogenous genomic regions, we used two endogenous normalizers viz. Actb and Gapdh. and show tissue specific methylation enrichments of the genes H19 in old (30 mo-old) mice vs. young (3 mo-old) mice and in middle age mice (17 mo-old) which were allowed to eat ad libitum or were subjected to 23% dietary restriction. In all these experiments, the enrichment of the H19 gene was about half than that of the HIST1H2BA/Hist1h2ba gene. This was expected since H19 is an imprinted geneCitation43 and the enrichment is directly proportional to the methylation content of the fragments.Citation29
For the mouse genomic DNA experiments, enrichment values were dependent not only on the methylation status of the region of interest but also on the biological variability in the methylation status of the endogenous genomic region chosen as normalizer. Comparison of results obtained for the two endogeneous normalizers Actb and Gapdh (, Panels C and D) clearly shows that differential methylation of the normalizers between tissues and treatments has a major impact on the results. To minimize these problems, we re-calculated enrichments for H19 and Hist1h2ba/HIST1H2BA using our spiked negative control as a normalizer. This provides an objective measure of the biological variability between individual samples within a group and enables direct comparison of the efficacy of the MeDIP process between experiments (between or within laboratories) if required.
To illustrate this issue, we present results from (1) an experiment in which the variation in estimates of relative enrichment was minimized by the use of the negative control instead of the endogenous normalizer ( and ) and (2) an experiment where apparently different enrichments between two tissues became non-significant when the negative control was used as a normalizer (). For , enrichment following MeDIP was calculated from qPCR data after WGA from old mice heart gDNA for the endogenous gene Hist1h2ba with Actb as the normalizer and compared with estimates of enrichment obtained after normalization with the negative control. As shown in Panel B, normalization with the negative control reduced considerably the coefficient of variation for the biological replicates; this CV better represents the biological variability due to the methylated endogenous region. , Panels C and D presents data on enrichments of the endogenous gene Hist1h2ba from heart and colon genomic DNA from young mice using either Actb or the spiked negative control as the normalizer. The apparently tissue-specific difference in Hist1h2ba methylation between colon and heart was no longer significant when the negative spike control was used as the normalizer. Constancy between spiked controls enrichments is confirmed in panel D, showing very similar degrees of enrichment post-WGA for DNA from the same samples. qPCR data in Panel C were confirmed either by BM direct sequencing or BM cloning and sequencing: heart samples showed lower methylation—especially at CpG no. 11 and 12—in the Hist1h2ba region analyzed (Panel E), and also lower methylation in the Actb region analyzed (Panels F and G). The actual Hist1h2ba methylation is the one obtained when normalizing with the negative control, while the opposite result was obtained when normalizing with Actb. Together, these data demonstrate that the methylation status of the endogenous region used as a normalizer may affect the estimates of enrichment obtained and, therefore, has the potential to confound interpretation of the results. We conclude that a better way to quantify biological variability in enrichment following MeDIP between samples within an experiment or for comparisons between different experiments is to calculate the enrichments using the spiked negative control as the comparator.
Figure 5. Panels A and B: normalization of endogenous gene enrichment by the negative control reduced experimental variability. A) Average fold enrichments for Hist1h2ba normalized with the endogenous region Actb (left column) or with the spiked negative control (right column) in old mice heart DNA. Data are mean ± SD, n = 3 biological replicates. Means are not significantly different (p = 0.41, Student’s t-test). B) Black bars show Coefficients of Variation (CV) related to estimates in panel A. Panels C and D: normalization of endogenous gene enrichment by the negative control reduced the impact of tissue-specific methylation of the normalizer gene. Panel C shows the comparison between gene specific enrichments for Hist1h2ba calculated with the endogenous normalizer Actb and with the negative control for young colon samples (white bars) and young heart samples (dashed bars). Panel D shows positive control/negative control enrichment post-WGA. Data are mean ± SD, n = 3 biological replicates. * p = 0.04, Student’s t-test. Panels E, F and G: validation of Panel C results by BM sequencing. Panel E shows direct BM sequencing of murine Hist1h2ba; data are mean ± SD, n = 3 biological replicates. Panel F and G show aggregate representation of methylation data obtained by Bisulfite Modified (BM) cloning and sequencing for one colon and one heart sample, respectively. The bars indicate the percentage of clones that showed unmethylated (blue) or methylated (yellow) cytosines at the CpG sites analyzed (adapted from BiQ Analyzer v2.00).
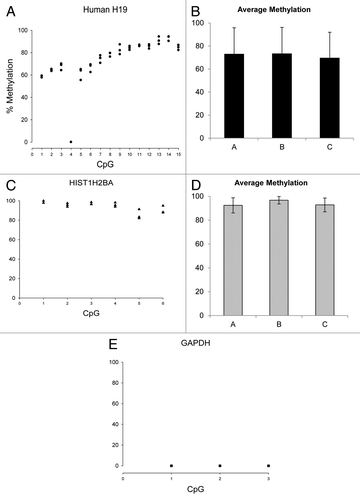
To further confirm our findings, we quantified a subset of CpG sites within the same endogenous regions analyzed by qPCR using BM DNA direct sequencing. Primers for the assays were designed with MethPrimer softwareCitation44 on 600 bp sequences (the average length of the fragments for Immunoprecipitation) centered on the described qPCR product (Table S3). As for the qPCR assays, we first assessed reproducibility and extent of error on 3 technical replicates—BM gDNA from human MRC5 cells—and then quantified the methylation content of one batch of biological replicates—young and old mouse colon BM gDNA.
shows percentage cytosine methylation for MRC5 fibroblasts in 3 technical replicates (Panels A, C and E). CVs range from 1.42% to 8.63% across 15 CpGs for H19 and from 1.26% to 5.85% across 6 CpG for HIST1H2BA. This statistic could not be calculated for GAPDH, as bisulfite modification of this region resulted in complete C to T conversion so there was no detectable cytosine methylation across the 3 CpG analyzed (data not shown). The mean percentage methylation for all the CpG sites examined was 71.90% for H19 (Panel B), 93.96% for HIST1H2BA (Panel D) and 0 for GAPDH (data not shown). These data provide evidence that, as expected, H19 was approximately hemi-methylated, HIST1H2BA was highly methylated and GAPDH was unmethylated. In addition, the ratio of mean methylation of HIST1H2BA/H19 is similar to the GAPDH normalized mean enrichment ratio of HIST1H2BA/H19 observed in our previous experiments (1.31 fold vs. 1.60 fold, respectively). This suggests that our method can be used as a semi-quantitative predictor for the calculation of methylation fold changes in samples to be compared, provided that a suitable normalizer gene (DNA domain) is used, i.e., one for which methylation is not affected by treatment.
Figure 6. Methylation quantification by direct bisulfite modified DNA sequencing of human MRC5 H19, HIST1H2BA and GAPDH regions (see Table S3 for primers and BM DNA sequences). Panels A, C and E Y axes present data measured for 3 independent technical replicates. Panels B and D present average methylation ± SD across the CpG sites analyzed.
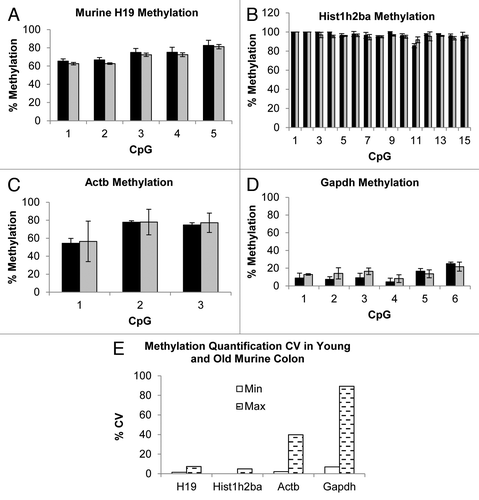
Panels A to D show percentage cytosine methylation in mouse colon. As for the human orthologous regions, mean percentage methylation in both young and old mice was about 70% in H19 across 5 CpGs and close to 100% in Hist1h2ba across 15 CpGs. Mean methylation of the housekeeping genes was approximately 70% across 3 CpGs for Actb and 13% across 6 CpGs for Gapdh, consistent with our enrichments results, which were higher when normalized using Gapdh (, Panels C and D). Panel E shows the minimum and the maximum percentage CV calculated on the CpG analyzed, which spanned from 0 (Hist1h2ba) to 89.28% (Gapdh), also consistent with data presented in , where Gapdh was the normalizer which showed the biggest SDs. These data indicate that there may be considerable inter-animal variation in methylation of conventional housekeeping genes and that researchers need to be alert to the possibility that their treatments may influence the methylation status of such genes. These observation reinforce the need for an external normalizer to provide a more precise and unbiased normalization of enrichment data, if a qPCR approach is used for quantification of methylation .
Figure 7. Methylation quantification by direct bisulfite modified DNA sequencing of H19, Hist1h2ba, Actb and Gapdh regions of murine Young (black bars) and Old (gray bars) colon samples (Panels A to D, see Table S3 for primers and BM DNA sequences). Error bars present SD calculated on each single CpGs measurements of 3 biological replicates. Panel E shows minimum (white bars) and maximum (dashed bars) percentage CV determined on all the investigated CpGs measurements.
Conclusion
In the present study, we developed standardization protocols and quality controls to assess the specificity, reproducibility and efficiency of the MeDIP procedure. By the use of “spiked” positive and negative controls, we provide an economical and fast method for quantifying success in enrichment of samples processed using the MeDIP technique. This method can be used: (1) to test the reproducibility, specificity and efficiency of MeDIP assays; (2) as a quality control in the screening of MeDIP processed samples prepared for hybridization to methylation microarrays; (3) to minimize the risk of bias when making comparisons in the extent of methylation between treatments/ conditions when there may be biological variation not only in the DNA regions of interest, but also in the endogenous regions used as normalizers and; (4) to normalize calculations of enrichment when the methylation status of DNA domains of interest is not known. We anticipate that the test protocols and quality controls described in this report will have utility when comparing results of MeDIP procedures between samples and experiments within laboratories and between laboratories.
Materials and Methods
Tissues and Cell Lines
Tissue samples were obtained from a long-established colony of the C57BL (ICRFa) mouse strain.Citation45 Hearts and colons were obtained from 3 mo old (Young) (n = 3) and 30 mo old (old) (n = 3) male mice. Livers were obtained from male mice aged 7 mo (n = 3), 17 mo (n = 3 fed ad libitum and n = 3 at 23% of dietary restriction for 3 mo before sample collection) and 32 mo (n = 3). Tissue samples were frozen in liquid nitrogen immediately after collection and stored at -80°C until use.
MRC5 human fibroblasts were cultured at 37°C in 5% CO2 atmosphere in DMEM medium (Sigma Aldrich) supplemented with 10% FBS, 100 U/mL Penicillin and 100 µg/mL Streptomycin. Cells were harvested at 100% confluence after 30 population doublings. Cell pellets were washed 3 times with PBS and stored at -80°C until use.
DNA Extraction
Tissue/cell pellets were digested overnight at 55°C in 50mM TRIS-HCl, 12.5mM EDTA, 0.5% SDS, pH = 8 and 0.03 U/ µL Proteinase K (Fermentas). Nucleic acids were isolated with a mixture 24:1 of Chloroform: Isoamyl Alcohol (Sigma Aldrich) and, after RNA digestion (RNase A/T1 mix, Fermentas), DNA was precipitated with isopropanol and resuspended in 50 µL of TE buffer. Liver and heart samples were washed with Red Blood Cell Lysis solution (QIAgen) before tissue digestion to eliminate trace amounts of blood. 200 ng DNA was loaded on 1% agarose gel with 0.4 µg/mL of Ethidium Bromide to check for integrity.
Preparation of DNA control samples
Positive (methylated) control DNA and negative (unmethylated) control DNA were prepared by PCR from lambda DNA dam- dcm- (Fermentas). Reactions were set up with 10 ng template DNA, 500 nM final primers concentration (primer pair 1, see ) and Fast Taq Mix (Fermentas). PCR conditions were as follows: 95°C for 1 min; 95°C for 1 sec and 60°C for 15 sec for 30 cycles; 72°C for 10 sec, in a total volume of 20 µL. PCR products were purified with silica columns (Fermentas). To prepare the fully methylated positive control, Lambda DNA Fragment A was methylated with M.SssI (New England BioLabs) according to the manufacturer’s instructions and re-purified with silica columns (Fermentas). The methylated positive control and unmethylated negative control were stored as single-use aliquots at -20°C until required.
Testing controls (Unmethylated and Methylated DNA fragments) by methylation sensitive restriction enzyme assays
DNA digestion reactions were set up with 200 ng DNA and 1 µL of each enzyme (AciI, MspI or HpaII FastDigest - purchased from Fermentas) in a total volume of 20 µL and incubated for 1 h at 37°C. Reaction products were loaded on 2% MetaPhor high resolution agarose (Lonza) gel with 0.4 µg/mL of Ethidium Bromide.
Fragmentation of sample DNA by sonication
Ten µg gDNA was diluted to 450 µL with TE buffer and sonicated in cold water for 2 min at 20% pulse and 5 Volts of power output with a Ultrasonic Homogenizer 4710 Series (Cole-Parmer Instrument Co.). Extent of sonication was assessed by loading 15 µL sample on 1% agarose gel with 0.4 µg/mL of Ethidium Bromide. Successfully fragmented DNA samples were concentrated with silica columns (Zymo Research) to 50 µL in TE buffer.
Preparation of Methylated DNA by ImmunoPrecipitation (MeDIP)
Reaction mixtures were prepared by adding 40 ng positive control and 40 ng negative control to 4.4 µg of sonicated gDNA in a total volume of 495 µL of TE buffer. After 10’ of denaturation at 95°C, samples were cooled in ice for 10’ and 1/10 of the reaction volume was stored as Input at 4°C. Immunoprecipitation reactions were performed as described previouslyCitation29 with the following modifications: we used 12 µg 5-methylcytidine antibody (Eurogentec), and 48 µL of Dynabeads M-280 Sheep anti-mouse IgG (Invitrogen). The denaturing elution from the beads with Proteinase K was performed at 50°C for 2 h and eluates purified with silica columns (Zymo Research) in a final volume of 15 µL of TE buffer.
Whole Genome Amplification (WGA) and assessment of WGA bias
WGA2 kit (Sigma) was used following the manufacturer’s instructions, using 20 ng of template DNA and omitting the fragmentation step. Products of the WGA reactions were cleaned up with silica columns (Sigma) and elutions were performed with 50 µL water. For the assessment of WGA length related bias, Lambda DNA fragments of different length were prepared by PCR using HotStart MasterMix (QIAgen),10 ng of template, Primers Pairs 1 listed in and Table S1 at a final concentration of 300 nM each and the following reaction conditions: 95°C for 15 min; 95°C for 30 sec and 60°C for 30 sec and 72°C for 1 min for 30 cycles; 72°C for 10 min. Total volume was 25 µL. PCR products were purified with silica columns (Fermentas) from 1% agarose gel with 0.4 µg/mL of Ethidium Bromide and spiked in gDNA at a ratio 1:100. Spiked samples were then processed for WGA as indicated above and 25 pg of each pre-WGA and post-WGA reaction was tested by qPCR with primer pairs 2 listed in and Table S1.
qPCR Assays
To design and perform all qPCR assays we followed the Minimum Information for Publication of Quantitative Real-Time PCR Experiments GuidelinesCitation46 (see Table S4, Fig. S1 and Fig. S2). All primer pairs used in qPCR assays were tested on at least three 10-fold dilutions of template for linearity of amplification and in 3 final concentrations (100 nM, 200 nM, 300 nM). Primers were designed to have an annealing temperature of 60°C and to produce amplicons of 90 to 150 bp. PCR reactions were performed in triplicate in a LightCycler 480 (Roche) with Maxima SYBR Green qPCR Master Mix (Fermentas) in the following conditions: 95°C for 10 min; 95°C for 15 sec and 60°C for 1 min for 45 cycles. Melting curves were obtained as follows: 95°C for 5 sec, 65°C for 1 min, gradual increase in temperature from 65°C to 97°C, with a ramp rate of 0.11°C/s. The acquisition of the fluorescence signal was set as a single reading during the annealing step and as a continuous reading during the melting curve. Primer concentrations were 300 nM for the endogenous gene assays and 100 nM for the spiked positive and negative control assays. Template quantities were 25 ng for the endogenous gene assays and 20 pg for the spiked control assays, each in a final volume of 25 µL. Ten µL of qPCR products were loaded on 1% agarose gel with 0.4 µg/mL of Ethidium Bromide to confirm the presence of a unique amplification band. Primer sequences are listed in , Table S1 and Table S2 and gel electrophoreses are shown in Figure S2.
qPCR products sequences confirmation
One µL of qPCR product of each endogenous region of interest and from one sample was subcloned in a pJET 1.2/blunt cloning vector using the CloneJET PCR cloning kit (Fermentas) and following the manufacturer’s instructions for sticky-end cloning. 50 µL aliquots of E. coli DH5α competent cells were heat-shock transformed with 5 µL of the ligation reactions (30 min in ice, 20 sec at 42°C and 2 min ice); 950 µL of SOC medium was added, followed by an incubation for 1 h at 37°C, 180 rpm. 1/10 of the transformation reactions was then plated on LB agar plates with 50 µg/mL Ampicillin and allowed to grow for 16h at 37°C. Five colonies for each subcloning were PCR screened for insertions with the vector primers provided in the kit (pJET1.2 FOR and pJET1.2 REV; among the positives, one was further confirmed by restriction digestion (BglII FastDigest, Fermentas) and then sequenced with T7 primer and pJET1.2 REV primer in an ABI 3130xl sequencer. The identities of the full-length PCR products were confirmed by BLAST analyses using the UCSC Genome Browser: we found 97.8% to 100% identity match (see Fig. S2).
Calculations of methylation enrichments following MeDIP preparation
The enrichment of methylated regions of DNA in the DNA fractions prepared by MeDIP was calculated from qPCR data using this formula:Citation35
Enrichment = (E target)∆Cq target (Input-MeDIP)/ (E reference)∆Cq reference (Input-MeDIP)
where “E” is the efficiency of amplification, “target” is the methylated region of interest and “reference” is the normalizer (a DNA region known, or expected, to be unmethylated in all samples).
Quantitative methylation analyses by direct sequencing
350 ng of genomic DNA was bisulfite converted using the EZ DNA Methylation-Gold kit (Zymo Research) according to the manufacturer’s instructions and eluted with 25 µL of the kit’s elution buffer. Two µL of the converted DNA was amplified with primers for Bisulfite Modified (BM) DNA, designed with MethPrimer softwareCitation44 (primers sequences and PCR conditions are listed in Table S3). Five µL of the PCR reactions was treated with 2 µL of Exo-SAP-IT (GE Healthcare) to eliminate excess primers and nucleotidesCitation47 (37°C, 15 min; 80°C, 15 min) and sequenced with the forward primer in an ABI 3130xl sequencer. Sequences were subject to visual quality control and areas with wide/overlapping peaks were excluded from subsequent analyses. A further quality control was performed with BiQ Analyzer softwareCitation48 to ensure a minimum of 80% identity match with the expected PCR product sequence and a minimum of 90% conversion rate of unmethylated cytosines to thymines. Cytosine methylation content was determined from the chromatograms applying the following formula:
% C Methylation = C peak height / (C peak height + T peak height) * 100.47
Quantitative Methylation Analyses by BM cloning and sequencing
when direct sequencing did not pass BiQ quality controls or sequencing chromatograms were not optimal, we cloned the BM PCR products as already described in the section “qPCR products sequences confirmation,” we purified DNA from 10 clones for each sample using the QIAgen Spin Miniprep Kit and sequenced them with T7 primer in an ABI 3130xl sequencer. We used BiQ Analyzer softwareCitation48 as previously described for quality control and sequences alignment and to produce the aggregated methylation data graphs presented in this study.
Additional material
Download Zip (588 KB)Acknowledgments
We thank Dr. Kerry Cameron, Adele Kitching, Liz Nicolson and Julie Wallace for assistance with mouse dissections, Dr. Caterina Grillo for helpful discussions and Dr. Mark Walker for allowing us extensive use of the Roche LightCycler 480.
Funding
This work was supported by the Centre for Integrated Systems Biology of Aging and Nutrition funded by the Biotechnology and Biological Sciences Research Council and by the Engineering and Physical Sciences Research Council [grant number BB/C0082001] and by the Biotechnology and Biological Sciences Research Council [grant numbers BB/G007993/1 and BB/I020748/1].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Illingworth R, Kerr A, Desousa D, Jørgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol 2008; 6:e22; http://dx.doi.org/10.1371/journal.pbio.0060022; PMID: 18232738
- Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics 2004; 20:1170 - 7; http://dx.doi.org/10.1093/bioinformatics/bth059; PMID: 14764558
- Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 2006; 103:1412 - 7; http://dx.doi.org/10.1073/pnas.0510310103; PMID: 16432200
- Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev 1991; 55:451 - 8; PMID: 1943996
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 66; http://dx.doi.org/10.1038/ng1990; PMID: 17334365
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001; 293:1089 - 93; http://dx.doi.org/10.1126/science.1063443; PMID: 11498579
- Richardson B. Impact of aging on DNA methylation. Ageing Res Rev 2003; 2:245 - 61; http://dx.doi.org/10.1016/S1568-1637(03)00010-2; PMID: 12726774
- Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev 2006; 20:1848 - 67; http://dx.doi.org/10.1101/gad.1422906; PMID: 16847345
- Spahn L, Barlow DP. An ICE pattern crystallizes. Nat Genet 2003; 35:11 - 2; http://dx.doi.org/10.1038/ng0903-11; PMID: 12947402
- Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci 2009; 66:596 - 612; http://dx.doi.org/10.1007/s00018-008-8432-4; PMID: 18985277
- Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet 1988; 78:151 - 5; http://dx.doi.org/10.1007/BF00278187; PMID: 3338800
- Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta 2007; 1775:138-62.
- Taby R, Issa JP. Cancer epigenetics. CA Cancer J Clin 2010; 60:376 - 92; http://dx.doi.org/10.3322/caac.20085; PMID: 20959400
- Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet 2007; 16:Spec No 1 R50 - 9; http://dx.doi.org/10.1093/hmg/ddm018; PMID: 17613547
- Mathers JC. Nutritional modulation of ageing: genomic and epigenetic approaches. Mech Ageing Dev 2006; 127:584 - 9; http://dx.doi.org/10.1016/j.mad.2006.01.018; PMID: 16513160
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008; 299:2877 - 83; http://dx.doi.org/10.1001/jama.299.24.2877; PMID: 18577732
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 2010; 20:332 - 40; http://dx.doi.org/10.1101/gr.096826.109; PMID: 20107151
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 2010; 20:434 - 9; http://dx.doi.org/10.1101/gr.103101.109; PMID: 20219945
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 2010; 20:440 - 6; http://dx.doi.org/10.1101/gr.103606.109; PMID: 20219944
- Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007; 8:286 - 98; http://dx.doi.org/10.1038/nrg2005; PMID: 17339880
- Kuo KC, McCune RA, Gehrke CW, Midgett R, Ehrlich M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res 1980; 8:4763 - 76; http://dx.doi.org/10.1093/nar/8.20.4763; PMID: 7003544
- Karimi M, Johansson S, Stach D, Corcoran M, Grandér D, Schalling M, et al. LUMA (LUminometric Methylation Assay)--a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res 2006; 312:1989 - 95; http://dx.doi.org/10.1016/j.yexcr.2006.03.006; PMID: 16624287
- Karimi M, Johansson S, Ekström TJ. Using LUMA: a Luminometric-based assay for global DNA-methylation. Epigenetics 2006; 1:45 - 8; http://dx.doi.org/10.4161/epi.1.1.2587; PMID: 17998810
- Olek A, Oswald J, Walter J. A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res 1996; 24:5064 - 6; http://dx.doi.org/10.1093/nar/24.24.5064; PMID: 9016686
- Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 1997; 25:2532 - 4; http://dx.doi.org/10.1093/nar/25.12.2532; PMID: 9171110
- Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93:9821 - 6; http://dx.doi.org/10.1073/pnas.93.18.9821; PMID: 8790415
- Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res 2000; 28:E32; http://dx.doi.org/10.1093/nar/28.8.e32; PMID: 10734209
- Uhlmann K, Brinckmann A, Toliat MR, Ritter H, Nürnberg P. Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis. Electrophoresis 2002; 23:4072 - 9; http://dx.doi.org/10.1002/elps.200290023; PMID: 12481262
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853 - 62; http://dx.doi.org/10.1038/ng1598; PMID: 16007088
- Klagsbrun M. An evolutionary study of the methylation of transfer and ribosomal ribonucleic acid in prokaryote and eukaryote organisms. J Biol Chem 1973; 248:2612 - 20; PMID: 4633356
- Karijolich J, Kantartzis A, Yu YT. Quantitative analysis of RNA modifications. Methods Mol Biol 2010; 629:21 - 32; http://dx.doi.org/10.1007/978-1-60761-657-3_2; PMID: 20387140
- Motorin Y, Lyko F, Helm M. 5-methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res 2010; 38:1415 - 30; http://dx.doi.org/10.1093/nar/gkp1117; PMID: 20007150
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, et al. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res; 39:D195-201.
- Butcher LM, Beck S. AutoMeDIP-seq: a high-throughput, whole genome, DNA methylation assay. Methods 2010; 52:223 - 31; http://dx.doi.org/10.1016/j.ymeth.2010.04.003; PMID: 20385236
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; http://dx.doi.org/10.1093/nar/29.9.e45; PMID: 11328886
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 2008; 26:779 - 85; http://dx.doi.org/10.1038/nbt1414; PMID: 18612301
- Feber A, Wilson GA, Zhang L, Presneau N, Idowu B, Down TA, et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome Res 2011; 21:515 - 24; http://dx.doi.org/10.1101/gr.109678.110; PMID: 21324880
- Lasken RS, Egholm M. Whole genome amplification: abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol 2003; 21:531 - 5; http://dx.doi.org/10.1016/j.tibtech.2003.09.010; PMID: 14624861
- Choi YC, Chae CB. DNA hypomethylation and germ cell-specific expression of testis-specific H2B histone gene. J Biol Chem 1991; 266:20504 - 11; PMID: 1718964
- Choi YC, Gu W, Hecht NB, Feinberg AP, Chae CB. Molecular cloning of mouse somatic and testis-specific H2B histone genes containing a methylated CpG island. DNA Cell Biol 1996; 15:495 - 504; http://dx.doi.org/10.1089/dna.1996.15.495; PMID: 8672246
- Tremblay KD, Duran KL, Bartolomei MS. A 5′ 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol Cell Biol 1997; 17:4322 - 9; PMID: 9234689
- Weber M, Milligan L, Delalbre A, Antoine E, Brunel C, Cathala G, et al. Extensive tissue-specific variation of allelic methylation in the Igf2 gene during mouse fetal development: relation to expression and imprinting. Mech Dev 2001; 101:133 - 41; http://dx.doi.org/10.1016/S0925-4773(00)00573-6; PMID: 11231066
- Gao ZH, Suppola S, Liu J, Heikkilä P, Jänne J, Voutilainen R. Association of H19 promoter methylation with the expression of H19 and IGF-II genes in adrenocortical tumors. J Clin Endocrinol Metab 2002; 87:1170 - 6; http://dx.doi.org/10.1210/jc.87.3.1170; PMID: 11889182
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18:1427 - 31; http://dx.doi.org/10.1093/bioinformatics/18.11.1427; PMID: 12424112
- Rowlatt C, Chesterman FC, Sheriff MU. Lifespan, age changes and tumour incidence in an ageing C57BL mouse colony. Lab Anim 1976; 10:419 - 42; http://dx.doi.org/10.1258/002367776780956917; PMID: 979138
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009; 55:611 - 22; http://dx.doi.org/10.1373/clinchem.2008.112797; PMID: 19246619
- Jiang M, Zhang Y, Fei J, Chang X, Fan W, Qian X, et al. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab Invest 2010; 90:282 - 90; http://dx.doi.org/10.1038/labinvest.2009.132; PMID: 20010852
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 2005; 21:4067 - 8; http://dx.doi.org/10.1093/bioinformatics/bti652; PMID: 16141249