Abstract
DNA methylation of CpGs located in two types of repetitive elements—LINE1 (L1) and Alu—is used to assess “global” changes in DNA methylation in studies of human disease and environmental exposure. L1 and Alu contribute close to 30% of all base pairs in the human genome and transposition of repetitive elements is repressed through DNA methylation. Few studies have investigated whether repetitive element DNA methylation is associated with DNA methylation at other genomic regions, or the biological and technical factors that influence potential associations. Here, we assess L1 and Alu DNA methylation by Pyrosequencing of consensus sequences and using subsets of probes included in the Illumina Infinium HumanMethylation27 BeadChip array. We show that evolutionary age and assay method affect the assessment of repetitive element DNA methylation. Additionally, we compare Pyrosequencing results for repetitive elements to average DNA methylation of CpG islands, as assessed by array probes classified into strong, weak and non-islands. We demonstrate that each of these dispersed sequences exhibits different patterns of tissue-specific DNA methylation. Correlation of DNA methylation suggests an association between L1 and weak CpG island DNA methylation in some of the tissues examined. We caution, however, that L1, Alu and CpG island DNA methylation are distinct measures of dispersed DNA methylation and one should not be used in lieu of another. Analysis of DNA methylation data is complex and assays may be influenced by environment and pathology in different or complementary ways.
Introduction
The theory that the environment can affect gene expression has gained much attention in recent years. This has led to widespread interest in assessing the effects of environmental exposures on epigenetic changes, including DNA methylation, at targeted loci as well as across the genome. Repetitive elements (REs) have been used as a surrogate to measure “global” changes in DNA methylationCitation1 associated with diverse factors including nutrition,Citation2 pollutantsCitation3,Citation4 and toxin exposure.Citation5 Several approaches can be used to study the same families of REs, and each method may yield varying results depending on the population of elements that is analyzed. The degree to which RE DNA methylation relates to DNA methylation at other genomic sequences and how different measures are influenced by biological and technical variables remains unclear.
Although there are numerous methods for measuring DNA methylation across the genome, one of the most widely used is high performance liquid chromatography (HPLC). Using HPLC, individual nucleotides are counted and a total measure of 5-methyl-cytosine (5-mC) content is obtained.Citation6 5-mC-specific antibody kits have been developed as a less expensive and less labor intensive alternative to HPLC.Citation7, Citation8 While both HPLC and antibody kits provide a measure of average global methylation, the distribution of 5-mCs within the genome cannot be determined with these methods. Alternatively, array-based methods sacrifice some genomic coverage for the ability to detect the specific location of 5-mCs. Methylated DNA immunoprecipitation (MeDIP) captures methylated fragments of DNA with a 5-mC-specific antibody and is used in combination with a microarray for comparison against a differentially labeled control.Citation9 Another array-based method, the Illumina Infinium HumanMethylation27 BeadChip array, allows single-nucleotide resolution of the DNA methylation status of 27,578 CpG sites in over 14,000 gene promoters.Citation10
DNA methylation of two RE families, LINE1 (L1) and Alu, has recently been used to assess genome-wide DNA methylation because L1 and Alu sequences account for close to 30% of the total number of base pairs in the human genome;Citation11 about 12% of all CpG dinucleotides fall within L1s while about 25% fall within Alu sequences.Citation12,Citation13 Alu and L1 transposable elements integrated into the ancestral genome more than 80 and 150 million years ago respectively, and over time, lineages have diverged significantly from the original DNA sequences.Citation14 Alu and L1s amplify by a copy-and-paste mechanism that reverse transcribes the repeat sequence into a new location.Citation11 The reverse transcriptase machinery is not robust: whereas the short Alu sequences of about 300 bps can be entirely transcribed,Citation15 full L1s are much longer (up to about 6,000 bps) and thus are 5′ truncated at many insertion sites.Citation16 Phylogenetic analyses divide the existing 500,000 copies of L1 and 1,000,000 copies of AluCitation14 each into three large subfamilies based on evolutionary age.Citation15,Citation17,Citation18 REs can have diverse effects on local genomic environment including altering gene expression and acting as sites for crossover, leading to duplications or deletions (reviewed in Kazazian, 2004Citation19). Full-length L1 elements contain a sense and antisense promoter close to their 5′ end, in addition to a sense promoter in the 3′UTRCitation20 that can act as alternative promoters.Citation21 It is hypothesized that the host genome methylates RE DNACitation9,Citation22 as a defense mechanism to limit detrimental transcription. However, these DNA methylation patterns differ based on the evolutionary age of the RE examined.Citation23 Given their copy number and the diversity of subfamily sequences, it is unlikely that different techniques used to measure RE DNA methylation, such as Pyrosequencing,Citation1,Citation24 MethyLightCitation25 and COBRACitation1 measure the same population of Alu or L1 elements.
There is evidence that REs are particularly prone to changes in DNA methylation with exposure to environmental toxins. In mice, feeding mothers the xenoestrogen bisphenol A (BPA) during pregnancy results in hypomethylation of the RE upstream of the Avy geneCitation26 and a coordinated change in coat color in the offspring.Citation27 Furthermore, supplying the mother with a methyl donor-rich diet partially counteracts the effects of BPA-induced hypomethylation.Citation26 In humans, several studies have reported small reductions in L1 and/or Alu DNA methylation in adults exposed to benzene,Citation28 particulate air pollution,Citation3,Citation29 DDTCitation4 and PCBs.Citation4
Since REs are present throughout the genome and sensitive to environmental exposures, measuring the DNA methylation status of Alu and L1 sequences is an attractive method to rapidly and economically assess the DNA methylation status of many CpGs.Citation1 There is however, little evidence in direct support of L1 and Alu DNA methylation as surrogates for genome-wide DNA methylationCitation25,Citation30 or DNA methylation of other groups of genomic sequences. The self-replication of REs in addition to their evolutionary age and parasitic relationship with the human genome makes L1 and Alu DNA methylation particularly interesting yet difficult to study. In order to design and compare experiments, there is a need to understand what L1 and Alu DNA methylation assays measure, how RE DNA methylation relates to other DNA methylation assays and the conditions that influence L1 and Alu assessment.
In this study, we use the term representative dispersed sequences (ReDS) to describe subsets of five targets—L1, Alu, strong CpG islands, weak CpG islands and non-islands—which were examined to determine “dispersed DNA methylation” in a set of placental chorionic villi at three gestational ages (1st trimester villi, 2nd trimester villi and term villi) and in four somatic tissues (fetal brain, fetal kidney, fetal muscle and adult blood; ). Alu and L1 DNA methylation was assessed by Pyrosequencing of consensus sequences in addition to examining groups of probes on the Illumina Infinium HumanMethylation27 BeadChip array that map to REs. DNA methylation of three categories of CpG islands was also examined using probes from the Illumina Infinium HumanMethylation27 BeadChip array: (1) strong island probes (map to high density CpG islands); (2) weak island probes (map to intermediate density CpG islands) and; (3) non-island probes (map to CpGs outside of islands) (see Methods for a full description of each assay).Citation9 Assessment of RE DNA methylation was compared between assays and correlation of DNA methylation was assessed between the five ReDS. Our analyses have highlighted that DNA methylation at CpG sites in L1 and Alu generally follow patterns distinct from other genomic sequences in the control tissues examined here.
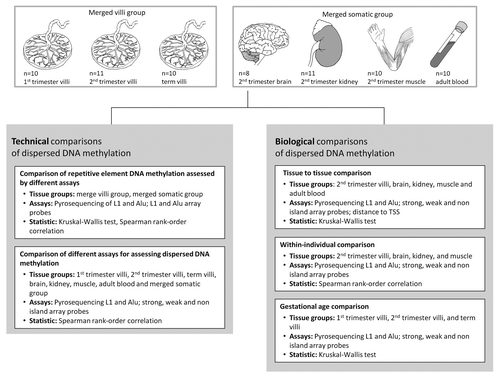
Figure 1. Schematic of analyses performed.
Results
Evolutionary age and assay method affect the assessment of L1 and Alu DNA methylation
Characteristics that may bias the assessment of RE DNA methylation include evolutionary age,Citation13,Citation23 genomic locationCitation13,Citation31 and assay method. DNA methylation of L1 and Alu sequences assessed by Pyrosequencing was compared with subsets of probes from the Illumina Infinium HumanMethylation27 BeadChip array that we identified as mapping to REs. Subfamily consensus sequences are used to design L1 and Alu Pyrosequencing primers and thus multiple primer sets are available for each family. Here, we used a primer set that measures four CpGs in the 5′ CpG island promoter of the L1H consensus sequence and three CpGs in the body of the AluSx consensus sequence, since these primer sets have been widely used in recent years.Citation1,Citation3,Citation4,Citation28,Citation29,Citation32 While there are about 130,000 copies of AluSx in the human genome,Citation15 there are only about 1,200 copies of L1H,Citation33 and up to 70% of these are expected to be 5′ truncated.Citation16 Thus, the L1 and Alu Pyrosequencing assays sample only a small portion of each subfamily and are only representative of these dispersed sequences. RE array probes cover a variety of evolutionary age groups but, due to the design of the array, the population we could examine was biased toward REs incorporated into gene promoters. A variable number of array probes mapped to each of the Alu and L1 subfamilies: old Alu (AluJ; n = 153), intermediate Alu (AluS; n = 392), young Alu (AluY; n = 78) and old L1 (L1M; n = 192), intermediate L1 (L1P; n = 26), young L1 (L1H; n = 4). This sample represents approximately 0.52% of all AluJ, 0.11% of all AluS, 0.08% of all AluY and 0.23% of all L1H, 0.01% of all L1P and 0.03% of all L1M.
Analyses of element age were conducted in two tissue groups: (1) merged villi (placental villi from all three gestational ages) and (2) merged somatic tissues (fetal brain, fetal kidney, fetal muscle and adult blood), since DNA methylation of each subfamily of Alu or L1 did not differ between the tissues included in each of these groups (data not shown). There was a significant trend for hypomethylation of older L1 and Alu subfamilies in comparison to intermediate and young in both the merged villi and somatic groups (). We next compared the level of RE DNA methylation assessed by Pyrosequencing to array probe assessment. After correction for multiple comparisons, mean L1 methylation by Pyrosequencing was not different from L1H methylation assessed by array probes in the merged villi group (). All other comparisons of DNA methylation between L1/Alu Pyrosequencing and array probes were significantly different in both tissue groups (, all p < 0.001). Assessment by Pyrosequencing of L1 DNA methylation was correlated with array results in the somatic tissue group (L1M, r = 0.55; L1P, r = 0.58; both p < 0.0001) but in neither tissue group for Alu methylation. Thus, DNA methylation of REs is dependent on both evolutionary age and assay method.
Figure 2. The assessment of Alu and L1 DNA methylation is affected by evolutionary age and assay type. DNA methylation of (A) Alu and (B) L1 was assessed in two groups: merged villi (n = 31; 1st trimester, 2nd trimester and term villi) and merged somatic tissues (n = 39; brain, kidney, muscle, blood) using Pyrosequencing and probes from the Illumina Infinium HumanMethylation27 BeadChip array. Array probes that mapped to REs were divided into three age groups based on evolutionary emergence: old Alu (AluJ; n = 113), intermediate Alu (AluS; n = 272), young Alu (AluY; n = 58) and old L1 (L1M; n = 24), intermediate L1 (L1P; n = 160), young L1 (L1H; n = 4). There was a trend for increased DNA methylation from the old to young Alu and L1 measured by the array. Alu and L1 DNA methylation as assessed by Pyrosequencing was significantly different from each age group measured by the array, except when comparing L1 by Pyrosequencing to young L1s in the merged villi group. Significance is indicated by *p < 0.05, **p < 0.001, ***p < 0.0001
Evidence for association of weak island probe methylation with L1 and non-island probe methylation
Correlation analyses were performed to determine how L1 and Alu DNA methylation assessed by Pyrosequencing compare with each other and to DNA methylation of other ReDS. Using average DNA methylation assessed by the array, samples clustered most strongly by placental vs. somatic origin (Fig. S1), and then by tissue type; thus, inter-method comparisons were conducted within each somatic tissue (fetal brain, kidney and muscle and adult blood) and then collectively in the merged somatic group. Gestational age was previously found to have a significant effect on villi DNA methylation,Citation34,Citation35 therefore, inter-method comparisons of villi were performed within each gestational age group. RE DNA methylation was compared with three groups of Illumina Infinium HumanMethylation27 BeadChip array probes categorized based on CpG island density: strong, weak and non-island probes. Table S1 summarizes average DNA methylation by tissue group for each of the five ReDS. There were no significant differences in DNA methylation by sex for any tissue or for any ReDS; thus, males and females were considered together for all analyses (Fig. S2).
L1 and weak island probe methylation were correlated in 2nd trimester villi (; r = 0.68, p = 0.025) and in brain (; r = 0.88, p = 0.007); however, these correlations did not withstand correction for multiple comparisons. Weak and non-island probe methylation were significantly correlated after correction for multiple comparisons in 1st trimester villi (; r = 0.94, p < 0.001) and 2nd trimester villi (; r = 0.80, p = 0.005). In the merged somatic group, weak island probe methylation was also correlated with L1 DNA methylation (; r = 0.48, p = 0.002) and with non-island probe methylation (; r = 0.91, p < 0.0001). L1 and Alu DNA methylation were correlated in 1st trimester villi (; r = 0.64, p = 0.054) and in brain (; r = 0.74, p = 0.046), but neither of these was significant after correction for multiple comparisons. Detailed correlation analyses within each tissue can be found in Figures S3–10. Since the correlation of DNA methylation between weak islands and non-islands in addition to weak islands and L1s was present in several individual tissues and the merged somatic group, the association between these two pairs of ReDS may be more than inter-individual variation. However, the inconsistency of correlation between DNA methylation of the other pairs of ReDS suggests that each measure targets a genomic sequence with different trends in DNA methylation.
Table 1. Spearman correlation of DNA methylation at five ReDS
Alternative dispersed DNA methylation assays each produce a distinct tissue-specific DNA methylation profile
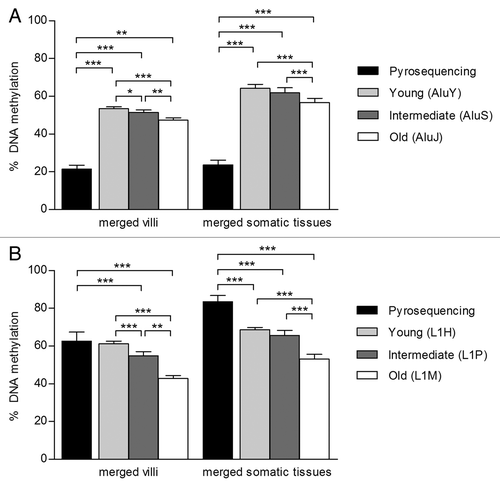
The five ReDS examined here are functionally different and thus trends in tissue-specific DNA methylation at these regions may also be distinct. Second trimester tissues (villi, brain, kidney and muscle) and adult blood were used to compare tissue-specific DNA methylation at each ReDS (). After correction for multiple comparisons, there were no significant tissue-to-tissue differences in average Alu DNA methylation (). However, average L1 DNA methylation in villi (62.52% ± 4.60) was significantly lower than in somatic tissues (; brain, 84.89% ± 3.32; muscle, 82.82% ± 2.44 and blood, 85.79% ± 1.92; all p < 0.001). Reduced average non-island probe methylation was also observed in villi (49.60% ± 1.20) compared with somatic tissues (; brain, 60.89% ± 1.48; kidney, 57.91% ± 0.93 and blood, 63.64% ± 0.58; all p < 0.001) and at weak island probes (; villi, 37.44% ± 0.65 vs. brain, 41.31% ± 0.49; kidney 38.83% ± 0.47 and blood, 43.53% ± 0.36; all p < 0.001). Interestingly, strong island DNA methylation in villi (11.21% ± 0.49) was greater than in other tissues (; muscle, 9.83% ± 0.32 and kidney, 9.80% ± 0.47; all p < 0.0001), although these small differences in DNA methylation may not be biologically significant. The similarity in the patterns of tissue-specific DNA methylation assessed by L1, weak island and non-island probe methylation further suggest that these ReDS may follow similar trends in DNA methylation.
Figure 3. Five ReDS exhibit different tissue patterns of DNA methylation. DNA methylation in villi (n = 11), brain (n = 8), kidney (n = 11) and muscle (n = 10) from 2nd trimester fetuses and adult blood (n = 10) was measured using (A) % Alu, (B) % L1, (C) strong island probes, (D) weak island probes and (E) non-island probes. L1, weak island and non-island probe methylation were most variable tissue to tissue. Villi DNA methylation was significantly reduced compared with most other somatic tissues at L1, weak island and non-island probes. However villi DNA methylation was significantly increased compared with kidney and muscle at strong island probes. Significance is indicated by *p < 0.05, **p < 0.001, ***p < 0.0001.
Distance to transcription start site is associated with promoter CpG island density and distinct trends in DNA methylation
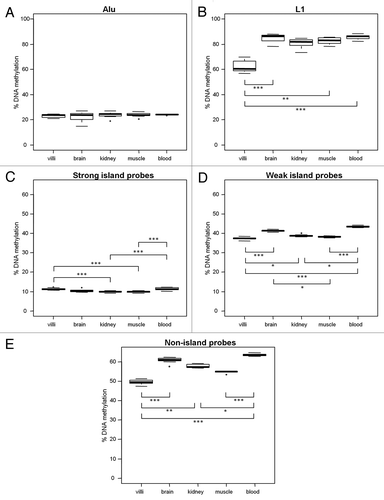
Since 98% of the array probes examined in the strong, weak and non-island groups were within 1500 bps from a known transcription start site (TSS), they are considered to be in gene promoters. We investigated whether probe distance to a TSS within each of the CpG island groups affected average DNA methylation. We present here the data from the merged somatic tissues; however, merged villi yielded similar results. Probes were binned into six 500 bp windows based on distance to nearest TSS: (1) -1500 to -1001 bps (n = 575), (2) -1000 to -501 bps (n = 1,989), (3) -500 to 0 bps (n = 8,628), (4) 1 to 500 bps (n = 8,707), (5) 501 to 1000 bps (n = 2,036) and (6) 1001 to 1500 bps (n = 558) (). DNA methylation was significantly different between strong, weak and non island CpGs within each bin (p<0.0001) but was not dependent on probe direction to TSS (upstream vs. downstream, p = 0.73); thus, direction to TSS was not considered in further analyses. We next assessed tissue-specific patterns of DNA methylation in 2nd trimester tissues and adult blood for probes of each CpG island density within each of the three TSS bins: (1) ± 0 to 500 bps, (2) ± 501 to 1000 bps and (3) ± 1001 to 1500 bps. Average DNA methylation of tissues followed the same rank-order within each of the three TSS groups for strong, weak, and non-island probes (data shown for strong islands (). These patterns were also the same as those observed when probes were not separated by distance to closest TSS (Fig. 3C, D and E). Between-tissue differences in strong-islands were largest at CpGs distal to a TSS (; villi 16.91% ± 0.7 vs. kidney 15.00% ± 0.63, p p<0.0001; blood 18.10% ± 0.74, vs. brain 16.13% ± 0.64, kidney 15.00% ± 0.63 and muscle 16.20% ± 0.42, all p<0.05) and smallest at CpGs close to TSS (Fig. 4B; villi 9.63% ± 0.67 vs. kidney 8.27%± 0.46, and muscle 8.10% ± 0.32, all p<0.001).To test for an association between distance to nearest TSS and CpG island density, we compared the observed to expected number of array probes in each of the three TSS bins (Fig. S11). There was an overrepresentation of strong island probes and underrepresentation of non-island probes close to TSS (± 0 to 500 bps). Conversely, there was an overrepresentation of non-island probes and an underrepresentation of strong island probes distal to TSS (± 1001 to 1500 bps). Taken together, these results imply that within promoters, distance to nearest TSS is linked to CpG island density, which influences DNA methylation of promoters.
Figure 4. Distance to transcription start site (TSS) influences methylation of probes in promoters. 98% of probes on the Illumina Infinium HumanMethylation27 BeadChip array were within 1500 bps of a known gene TSS. (A) In the merged villi group, DNA methylation was analyzed by binning probes into six 500 bp windows around known TSS. There were significant differences in DNA methylation between probes in strong (●) weak (■) and non(▲) islands (p<0.0001) but direction to TSS had no significant effect on DNA methylation. DNA methylation of probes furthest from TSS (± 1000 to 1500 bps; 16.31% ± 2.26, 59.29% ± 5.39, 63.82% ± 3.6 for strong, weak and non islands respectively) was significantly higher than probes close to TSS (± 0 to 500 bps; 8.65% ± 0.77; 35.60% ± 2.16, 57.14 ± 3.33 for strong, weak and non island respectively, all p<0.001). Tissue differences in DNA methylation of 2nd trimester villi, muscle, kidney, brain and adult blood were investigated in three TSS bins in strong CpG islands: (B) ± 0 to 500 bps, (C) ± 501 to 1000 bps and (D) ± 1000 to 1500 bps. Significance is indicated by *p < 0.05, **p < 0.001, ***p < 0.0001.
Preliminary within-individual correlation of dispersed DNA methylation
After examining tissue-specific patterns of dispersed DNA methylation, we investigated whether there were within-individual trends at each of the five ReDS. This is of particular interest in studies of environmental exposure and pathological conditions, to determine if changes in DNA methylation are localized to one tissue or are more widespread. The 2nd trimester fetal samples including villi, brain, kidney and muscle were obtained from 12 fetuses and were thus used to investigate intra-individual DNA methylation. None of the ReDS was universally significantly correlated across all four tissues analyzed within an individual (Fig. S12A-E). However, weak island probe methylation was significantly correlated in villi and kidney (Fig. S12D; r = 0.70, p = 0.03) and villi and brain (Fig. S12D; r = 0.89, p = 0.01). Additionally, Alu DNA methylation was correlated in villi and kidney (Fig. S12A; r = 0.82, p = 0.01). Overall, we observed more positive than negative correlation coefficients (Fig. S12; 18 vs. 7). In particular, there were more comparisons with r > 0.5 than for r < -0.5 (Fig. S12; 8 vs. 1). These results suggest a general trend for correlation of DNA methylation between tissues; however, conclusions are limited by the small sample size.
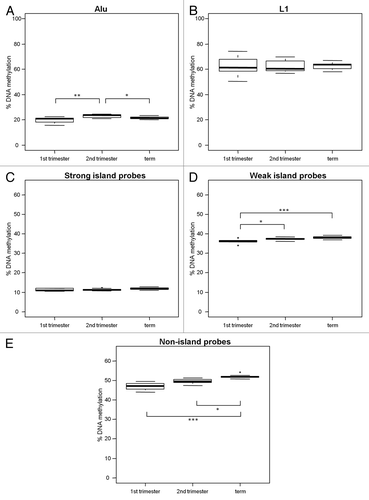
Increase of villi weak and non-island probe methylation throughout gestation
We previously reported an increase in villi gene promoter methylation throughout gestation using the Illumina Infinium HumanMethylation27 BeadChip array data for 1st trimester, 2nd trimester and term villi.Citation35 Here, we evaluated gestational age changes in villi DNA methylation with the addition of L1 and Alu DNA methylation, and further subdivision of array probes into strong, weak and non-islands (). After correction for multiple comparisons, there was a significant increase in non-island probe methylation from 1st trimester (46.99% ± 1.75) to term (52.01% ± 0.91) (; p < 0.0001) and weak island probe methylation from 1st trimester (36.12% ± 0.99) to term (38.10% ± 0.68) (; p < 0.0001) while strong island probe methylation was not altered between gestations (; 1st trimester, 11.15% ± 0.61; 2nd trimester, 11.21% ± 0.49 and term 11.82% ± 0.63). Mean L1 DNA methylation did not change throughout gestation, although variation did decrease (; 1st trimester, 62.69% ± 7.00; 2nd trimester, 62.52% ± 4.60 and term, 62.54% ± 2.78). Thus, villi gain in DNA methylation throughout gestation occurs at specific ReDS.
Figure 5. Increase in DNA methylation at weak and non-islands throughout gestation. DNA methylation in 1st trimester (n = 10), 2nd trimester (n = 11) and term (n = 10) placental villi was measured with (A) % Alu, (B) % L1, (C) strong island probes, (D) weak island probes and (E) non-island probes. There was no change in L1 or strong island methylation, but a notable increase in methylation at weak and non-islands throughout gestation. Significance is indicated by *p < 0.05, **p < 0.001, ***p < 0.0001.
Discussion
The phrase “global DNA methylation” has been widely and indiscriminately used in the literature to describe DNA methylation measured by a variety of techniques. Arguably, the only true global measures of DNA methylation assess genome-wide total 5-mC content. However, both L1 and Alu DNA methylation are commonly used as surrogates for “global DNA methylation,” implying that the status of the genome is being examined comprehensively. In this study, we examined representative members of five targets for DNA methylation (ReDS)—L1, Alu, strong CpG island promoters, weak CpG island promoters and non-island promoter CpGs—in placental villi, fetal organ tissues and adult blood. We have shown that the assessment of L1 and Alu DNA methylation is affected by assay method, evolutionary age composition of REs and tissue type. Additionally, distinct inter-tissue patterns of DNA methylation were observed at each of the ReDS. The subdivision of Illumina Infinium HumanMethylation27 BeadChip array probes into strong, weak and non-island probes, showed that the gestational age-related gain in villi DNA methylation observed in other studiesCitation35-Citation37 predominantly occurred at weak CpG island and non-island regions.
L1 and Alu sequences account for about 17% and 11% of the human genome, respectively,Citation14 and only a subset of these can be interrogated by any given technique. Although many studies use both L1 and Alu as surrogate measures for “global DNA methylation,” few have examined the question of whether L1 and Alu DNA methylation correlate with each other and with total 5-mC levels. Choi et al. reported a correlation of L1 with Alu DNA methylation in neuroendocrine tumors but not in control samples.Citation38 Given that REs may be sensitive sites in the genome for changes in DNA methylation, the correlation of L1 and Alu DNA methylation may be stronger under pathological conditions. This theory is supported by two additional studies in cancer cellsCitation39,Citation40 as well as our negative findings in control human tissues. Wang et al. found no correlation of total 5-mC, measured by Methylamp with mean L1 DNA methylation in human nervous tissue.Citation30 However, a study by Weisenberger et al. in blood identified a strong association of both L1 and Alu DNA methylation with total 5-mC content measured by HPLC.Citation25 Some of this study-to-study variation may be attributed to the use of different methods for measuring RE DNA methylation as well as total 5-mC content. Our comparison of RE DNA methylation obtained by Pyrosequencing vs. array probes suggests that the assessment of L1 and Alu DNA methylation is assay-dependent and thus may contribute to how these ReDS correlate with a genome-wide measure of 5-mC.
Using DNA methylation of different ReDS revealed that tissue to tissue DNA methylation was most variable at L1, non-islands and weak CpG islands and that the pattern of DNA methylation across tissues was similar at these three ReDS. Correlations between L1, weak CpG island and non-island methylation also suggest that these measures of dispersed DNA methylation may be associated with each other. Studies examining the genomic distribution of REs have demonstrated an enrichment of L1 in low GC content regions and on the X chromosome,Citation14 in addition to an underrepresentation near genes, where Alu elements are overrepresented.Citation41 L1s may be sites of de novo DNA methylation in the developing embryo from which DNA methylation silencing is spread TSS.Citation42 However, this spread into CpG island promoters may be buffered by Alus, which are enriched near TSSs associated with CpG islands.Citation31 We observed lower levels of DNA methylation in CpG island promoters close to TSS across all tissue types and increased DNA methylation at CpGs in island promoters further away from TSS. Kang et al. proposed a model of counteracting forces in the spread of DNA methylation toward promoters that leads to a transitional area of DNA methylation bordering CpG island promoters.Citation31 However, Kang et al. use a definition for CpG islands that falls in between the weak and strong CpG islands identified in our study. The spreading model of DNA methylation from L1 sequences suggests an explanation for the correlation we observed between L1 and weak CpG island DNA methylation.
Studies using HPLC have determined that the placenta has the lowest 5-mC content in comparison to other normal tissues.Citation36,Citation43,Citation44 Thus, the placenta is often described as hypomethylated compared with other fetal and adult tissues. Specific regions have also been shown to have decreased DNA methylation in the placenta, including Alu,Citation45 L1 and regions on the X chromosome.Citation46 Our study confirmed previous reports that L1s are hypomethylated in the placenta vs. other tissues. Some active L1 and Alu elements may play a functional role in the placenta, contributing to its invasive and proliferative properties. Placental-specific gene expression has been shown from both L1Citation21 and AluCitation47 sequences and may be more common in the placenta due to the lower levels of L1 and Alu DNA methylation. Syncytin is a classic example of another type of transposable element expressed exclusively in the placenta throughout gestationCitation48 and plays a critical role in the fusion of pluripotent cytotrophoblast into differentiated syncytiotrophoblast cells.Citation49 Interestingly, we found that there were not large differences in the DNA methylation of placenta compared with somatic tissues at strong CpG islands or close to TSS, in support of previous findings on the X chromosome.Citation46 Since L1 and Alu elements make up almost 30% of the genome,Citation14 REs may bias genome-wide tissue comparisons of DNA methylation based on total 5-mC content.
Although REs are widespread elements that can be rapidly and inexpensively assessed, our results suggest that L1s and Alus are sequences in the genome with distinct trends in DNA methylation. In fact, L1 DNA methylation may be more closely associated with weak CpG island methylation than “global DNA methylation.” Despite our findings, it should not be ruled out that under significant environmental changes or pathological conditions, DNA methylation of different ReDS may be more strongly associated and there may be greater intra-individual correlation. Conversely, DNA methylation of one type of dispersed sequence may be affected by a given condition while others are spared. Thus, the DNA methylation status of multiple ReDS are valuable measures to consider. We propose that L1 and Alu DNA methylation not be extrapolated to represent trends at other ReDS; they should be reported as a measure of methylation at CpG sites in the consensus sequence of specific REs. Additionally, we recommend that DNA methylation results only be compared when elements are measured by the same technique. The findings of our study reveal that the interpretation of dispersed DNA methylation data collected via a diverse range of assays is complex and the true power of these individual measurements and their relationship to each other should be scrutinized.
Methods
Sample collection
Samples were collected through the BC Women’s Hospital pathology department and previously described in Yuen et al.Citation50,Citation51 The study was approved by the ethics committees of the University of British Columbia and the Children’s and Women’s Health Centre of British Columbia (H04–70488, H06–70085). First trimester placenta (male n = 8, female n = 2) were obtained anonymously from elective terminations between 8–12 weeks gestation. Second trimester placenta (male n = 5, female n = 6) and fetal tissues (kidney, male n = 5, female n = 6; brain, male n = 4, female n = 4; muscle, male n = 4, female n = 6) were obtained from anonymous chromosomally normal pregnancies terminated for medical reasons such as premature rupture of membranes or placental abruption between 17–24 weeks gestation. Term placenta (38–41 weeks gestation, male n = 5, female n = 5) and adult female blood (n = 10) were samples used for previous studies of DNA methylation in the placenta.Citation34 After collection, a 1 cm3 piece was sampled from the fetal side of each placenta to avoid maternal decidual contamination. The amniotic membrane and chorion were removed and then DNA was extracted from one quarter of the piece of chorionic villi. A similar sized piece of each fetal tissue was also taken for DNA extraction. The remainder of each sample was stored at -80 C for future use. Extraction was performed by standard salt method for all samples and DNA was stored at -20 C.
Pyrosequencing
Methylation of L1 and Alu elements was measured by Pyrosequencing. 300 ng of genomic DNA was bisulfite converted using the EZ DNA Methylation Gold Kit (Zymo Research). L1 and Alu elements were amplified using published primer sets designed to complement the L1H and AluSx consensus sequences and cycling conditions used were as previously published.Citation28 PCR products were subsequently sequenced by a PyroMark MD system (Biotage). The DNA methylation status for each CpG dinucleotide was evaluated using PyroQ-CpG software (Biotage) and the average for all CpG sites was calculated as a percentage for each sample. Correlation of distinct bisulfite conversions for the L1 and Alu assays were r = 0.99 and r = 0.51 respectively. The lower Alu correlation is in part due to the small range of methylation values measured for the Alu assay. There was an average difference of only 1.6% methylation between Alu technical replicates.
Illumina Infinium HumanMethylation27 BeadChip array
All samples were run on the Illumina Infinium HumanMethylation27 BeadChip array. Data for villi, fetal tissues and blood samples were previously published in an analysis of gestational-age changes in DNA methylationCitation35 and for identification of novel differentially methylated regions between fetal tissues and blood.Citation51 Briefly, genomic DNA was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research), digested and then hybridized to the Inifinium HumanMethylation27 BeadChip array (Illumina). The Illumina Infinium HumanMethylation27 BeadChip array is a robust assay and technical replicates were highly correlated (r = 0.99). Using GenomeStudio 2008 1.0.5 software (Illumina), sex chromosome probes were removed from analysis to reduce sex-bias (n = 1,092). Poor quality probes (n = 32), defined as probes with detection p-value of > 0.05, in > 10% of samples were removed. Additionally we removed probes that were polymorphic at the target CpG (n = 263). Signal intensity output from genome studio was read in R (www.R-project.org) with the Bioconductor methylumi package to calculate an M value based on the ratio of the intensity of the methylated to unmethylated probes.Citation52 The lumi Bioconductor package was used to correct color biases from chip to chip and then a β value was calculated for each probe.
GpGIE2.0Citation53 was used to annotate the genome with the location, size and density of CpG islands based on Weber’s definition.Citation9 This annotation was intersected with the genomic location of array probes in Galaxy (http://galaxyproject.org/) to group probes into three categories based on the density of the CpG island in which they fell. Strong island probes (n = 14,391) were defined as those that mapped to CpG islands with > 55% GC content, > 0.75 observed/expected GC ratio and > 500 bps in length. Weak island probes (n = 2,786) were defined as those that mapped to CpG islands with > 50% GC content, > 0.48 observed/expected GC ratio and > 200 bps in length. Probes in weak islands that bordered strong islands (n = 2,229) were not included since they may act like CpG island shores and thus confound analyses. Non-island probes (n = 5,805) were all remaining probes. Probes that mapped to Alu (n = 623) and L1 (n = 222) were removed from CpG island groups and used for evolutionary age comparison of DNA methylation of REs. An estimate of the representation of all Alu and L1 sequences was obtained by dividing the number of probes on the array by the total number of sequences in each subfamily. The total number of Alu sequences was estimated based on supporting data in Price et al.Citation54 whereas the total number of L1 sequences was estimated base on counts from the UCSC RepeatMasker track (hg18).
Binning of probes into distance to nearest TSS was accomplished using distances provided in the Illumina Infinium HumanMethylation27 BeadChip array annotation file. Strong, weak and non-island probes were initially grouped separately into each of six non-overlapping bins: (1) -1500 to -1001 bps, (2) -1000 to -501 bps, (3) -500 to 0 bps, (4) 1 to 500 bps, (5) 501 to 1000 bps and (6) 1001 to 1500 bps. Average DNA methylation was calculated for probes in three groups based on CpG island density within each of the six TSS bin. Error bars give the standard deviation of this average between somatic tissue samples. Given that we found no difference in DNA methylation based on direction to TSS, probes were not separated into upstream/downstream in later TSS analyses.
Global DNA Methylation
Total 5-mC content of each sample was measured using the MethylFlash Methylated DNA Quantification Kit (Colorimetric) (Epigentek Group Inc.) following the manufacturerr’s protocol. As our results were variable and correlation of technical replicates was not significant, these data were not included in this publication.
Statistical Analyses
Comparison of methylation between tissue groups, gestational ages and RE evolutionary age was performed using the non-parametric ANOVA, Kruskal-Wallis test, followed by Dunn’s multiple comparison tests. Averages of DNA methylation are stated for tissue groups ± standard deviation. Spearman’s rank order correlation (r) was used to compare DNA methylation of different ReDS within each tissue group and within an individual, followed by Benjamini-Hochberg correction for multiple comparisons. A chi-square test was used to compare the observed vs. expected number of probes in each of the TSS bins. Statistics were performed in GraphPad Prism 5 (GraphPad Software, Inc.). Graphs were produced in R and GraphPad Prism 5. For box plots, boxes represent the interquartile range (IQR), whiskers the last data point within ± 1.5 x IQR, bars the median and dots outliers.
| Abbreviations: | ||
| Villi | = | placental chorionic villi |
| L1 | = | long interspersed nuclear element (LINE1) |
| 5mC | = | 5-methylcyotsine |
| RE | = | repetitive element |
| ReDS | = | representative dispersed sequences |
| TSS | = | transcription start site |
Additional material
Download Zip (2.4 MB)Acknowledgments
The authors would like to thank Ruby Jiang for technical assistance, Courtney Hanna and Dr. Kirsten Hogg for their constructive comments, Lucia Lam and Sarah Neumann for aid in microarray processing and analysis, Dr. Angela Devlin for use of the Biotage PyroMarkTM MD machine, Dr. Dixie Mager for useful conversations and Katarzyna Stepien for her figure illustrations. This work was supported by funds from the BC Clinical Genomics Network, a grant from the Canadian Institute for Health Research to WPR, and a training fellowship from the Child and Family Research Institute to EMP. MSK is a Scholar of the Michael Smith Foundation for Health Research and the Canadian Institute for Advanced Research. Sources of support include BC Clinical Genomics Network, Canadian Institute for Health Research and the Child and Family Research Institute
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Yang AS, Estécio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32:e38; http://dx.doi.org/10.1093/nar/gnh032; PMID: 14973332
- Fryer AA, Nafee TM, Ismail KMK, Carroll WD, Emes RD, Farrell WE. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics 2009; 4:394 - 8; http://dx.doi.org/10.4161/epi.4.6.9766; PMID: 19755846
- Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med 2009; 179:572 - 8; http://dx.doi.org/10.1164/rccm.200807-1097OC; PMID: 19136372
- Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect 2008; 116:1547 - 52; http://dx.doi.org/10.1289/ehp.11338; PMID: 19057709
- Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect 2010; 118:790 - 5; http://dx.doi.org/10.1289/ehp.0901429; PMID: 20064768
- Ramsahoye BH. Measurement of genome wide DNA methylation by reversed-phase high-performance liquid chromatography. Methods 2002; 27:156 - 61; http://dx.doi.org/10.1016/S1046-2023(02)00069-5; PMID: 12095275
- Li W, Liu M. Distribution of 5-hydroxymethylcytosine in different human tissues. J Nucleic Acids 2011; 2011:870726; http://dx.doi.org/10.4061/2011/870726; PMID: 21772996
- Chen X, Guo J, Lei Y, Zou J, Lu X, Bao Y, et al. Global DNA hypomethylation is associated with NTD-affected pregnancy: A case-control study. Birth Defects Res A Clin Mol Teratol 2010; 88:575 - 81; http://dx.doi.org/10.1002/bdra.20670; PMID: 20641100
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 66; http://dx.doi.org/10.1038/ng1990; PMID: 17334365
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009; 1:177 - 200; http://dx.doi.org/10.2217/epi.09.14; PMID: 22122642
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet 2009; 10:691 - 703; http://dx.doi.org/10.1038/nrg2640; PMID: 19763152
- Schmid CW. Alu: structure, origin, evolution, significance and function of one-tenth of human DNA. Prog Nucleic Acid Res Mol Biol 1996; 53:283 - 319; http://dx.doi.org/10.1016/S0079-6603(08)60148-8; PMID: 8650306
- Xie H, Wang M, Bonaldo MdeF, Smith C, Rajaram V, Goldman S, et al. High-throughput sequence-based epigenomic analysis of Alu repeats in human cerebellum. Nucleic Acids Res 2009; 37:4331 - 40; http://dx.doi.org/10.1093/nar/gkp393; PMID: 19458156
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al, International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921; http://dx.doi.org/10.1038/35057062; PMID: 11237011
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet 2002; 3:370 - 9; http://dx.doi.org/10.1038/nrg798; PMID: 11988762
- Buzdin A, Ustyugova S, Gogvadze E, Lebedev Y, Hunsmann G, Sverdlov E. Genome-wide targeted search for human specific and polymorphic L1 integrations. Hum Genet 2003; 112:527 - 33; PMID: 12601470
- Schmid CW. Human Alu subfamilies and their methylation revealed by blot hybridization. Nucleic Acids Res 1991; 19:5613 - 7; http://dx.doi.org/10.1093/nar/19.20.5613; PMID: 1945838
- Smit AF, Tóth G, Riggs AD, Jurka J. Ancestral, mammalian-wide subfamilies of LINE-1 repetitive sequences. J Mol Biol 1995; 246:401 - 17; http://dx.doi.org/10.1006/jmbi.1994.0095; PMID: 7877164
- Kazazian HH Jr.. Mobile elements: drivers of genome evolution. Science 2004; 303:1626 - 32; http://dx.doi.org/10.1126/science.1089670; PMID: 15016989
- Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 2009; 41:563 - 71; http://dx.doi.org/10.1038/ng.368; PMID: 19377475
- Mätlik K, Redik K, Speek M. L1 antisense promoter drives tissue-specific transcription of human genes. J Biomed Biotechnol 2006; 2006:71753; http://dx.doi.org/10.1155/JBB/2006/71753; PMID: 16877819
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853 - 62; http://dx.doi.org/10.1038/ng1598; PMID: 16007088
- Szpakowski S, Sun X, Lage JM, Dyer A, Rubinstein J, Kowalski D, et al. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 2009; 448:151 - 67; http://dx.doi.org/10.1016/j.gene.2009.08.006; PMID: 19699787
- Tost J, Dunker J, Gut IG. Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing. Biotechniques 2003; 35:152 - 6; PMID: 12866415
- Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res 2005; 33:6823 - 36; http://dx.doi.org/10.1093/nar/gki987; PMID: 16326863
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 2007; 104:13056 - 61; http://dx.doi.org/10.1073/pnas.0703739104; PMID: 17670942
- Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J 1998; 12:949 - 57; PMID: 9707167
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res 2007; 67:876 - 80; http://dx.doi.org/10.1158/0008-5472.CAN-06-2995; PMID: 17283117
- Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, et al. Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect 2009; 117:217 - 22; PMID: 19270791
- Wang L, Wang F, Guan J, Le J, Wu L, Zou J, et al. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am J Clin Nutr 2010; 91:1359 - 67; http://dx.doi.org/10.3945/ajcn.2009.28858; PMID: 20164316
- Kang MI, Rhyu MG, Kim YH, Jung YC, Hong SJ, Cho CS, et al. The length of CpG islands is associated with the distribution of Alu and L1 retroelements. Genomics 2006; 87:580 - 90; http://dx.doi.org/10.1016/j.ygeno.2006.01.002; PMID: 16488573
- Baccarelli A, Wright R, Bollati V, Litonjua A, Zanobetti A, Tarantini L, et al. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 2010; 21:819 - 28; http://dx.doi.org/10.1097/EDE.0b013e3181f20457; PMID: 20805753
- Ewing AD, Kazazian HH Jr.. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 2010; 20:1262 - 70; http://dx.doi.org/10.1101/gr.106419.110; PMID: 20488934
- Yuen RK, Peñaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet 2010; 18:1006 - 12; http://dx.doi.org/10.1038/ejhg.2010.63; PMID: 20442742
- Novakovic B, Yuen RK, Gordon L, Penaherrera MS, Sharkey A, Moffett A, et al. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genomics 2011; 12:529; http://dx.doi.org/10.1186/1471-2164-12-529; PMID: 22032438
- Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet 2004; 68:196 - 204; http://dx.doi.org/10.1046/j.1529-8817.2004.00081.x; PMID: 15180700
- Chavan-Gautam P, Sundrani D, Pisal H, Nimbargi V, Mehendale S, Joshi S. Gestation-dependent changes in human placental global DNA methylation levels. Mol Reprod Dev 2011; 78:150; http://dx.doi.org/10.1002/mrd.21296; PMID: 21425205
- Choi IS, Estecio MR, Nagano Y, Kim H, White JA, Yao JC, et al. Hypomethylation of LINE-1 and Alu in well-differentiated neuroendocrine tumors (pancreatic endocrine tumors and carcinoid tumors). Mod Pathol 2007; 20:802 - 10; http://dx.doi.org/10.1038/modpathol.3800825; PMID: 17483816
- Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, et al. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol 2007; 211:269 - 77; http://dx.doi.org/10.1002/path.2106; PMID: 17139617
- Richards KL, Zhang B, Baggerly KA, Colella S, Lang JC, Schuller DE, et al. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS One 2009; 4:e4941; http://dx.doi.org/10.1371/journal.pone.0004941; PMID: 19293934
- Medstrand P, van de Lagemaat LN, Mager DL. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res 2002; 12:1483 - 95; http://dx.doi.org/10.1101/gr.388902; PMID: 12368240
- Turker MS. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 2002; 21:5388 - 93; http://dx.doi.org/10.1038/sj.onc.1205599; PMID: 12154401
- Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 1982; 10:2709 - 21; http://dx.doi.org/10.1093/nar/10.8.2709; PMID: 7079182
- Novakovic B, Wong NC, Sibson M, Ng HK, Morley R, Manuelpillai U, et al. DNA methylation-mediated down-regulation of DNA methyltransferase-1 (DNMT1) is coincident with, but not essential for, global hypomethylation in human placenta. J Biol Chem 2010; 285:9583 - 93; http://dx.doi.org/10.1074/jbc.M109.064956; PMID: 20071334
- Hellmann-Blumberg U, Hintz MF, Gatewood JM, Schmid CW. Developmental differences in methylation of human Alu repeats. Mol Cell Biol 1993; 13:4523 - 30; PMID: 8336699
- Cotton AM, Avila L, Penaherrera MS, Affleck JG, Robinson WP, Brown CJ. Inactive X chromosome-specific reduction in placental DNA methylation. Hum Mol Genet 2009; 18:3544 - 52; http://dx.doi.org/10.1093/hmg/ddp299; PMID: 19586922
- Macaulay EC, Weeks RJ, Andrews S, Morison IM. Hypomethylation of functional retrotransposon-derived genes in the human placenta. Mamm Genome 2011; 22:722 - 35; http://dx.doi.org/10.1007/s00335-011-9355-1; PMID: 21874386
- Okahara G, Matsubara S, Oda T, Sugimoto J, Jinno Y, Kanaya F. Expression analyses of human endogenous retroviruses (HERVs): tissue-specific and developmental stage-dependent expression of HERVs. Genomics 2004; 84:982 - 90; http://dx.doi.org/10.1016/j.ygeno.2004.09.004; PMID: 15533715
- Frendo JL, Olivier D, Cheynet V, Blond JL, Bouton O, Vidaud M, et al. Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Mol Cell Biol 2003; 23:3566 - 74; http://dx.doi.org/10.1128/MCB.23.10.3566-3574.2003; PMID: 12724415
- Yuen RK, Neumann SM, Fok AK, Peñaherrera MS, McFadden DE, Robinson WP, et al. Extensive epigenetic reprogramming in human somatic tissues between fetus and adult. Epigenetics Chromatin 2011; 4:7; http://dx.doi.org/10.1186/1756-8935-4-7; PMID: 21545704
- Yuen RK, Jiang R, Peñaherrera MS, McFadden DE, Robinson WP. Genome-wide mapping of imprinted differentially methylated regions by DNA methylation profiling of human placentas from triploidies. Epigenetics Chromatin 2011; 4:10; http://dx.doi.org/10.1186/1756-8935-4-10; PMID: 21749726
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587; http://dx.doi.org/10.1186/1471-2105-11-587; PMID: 21118553
- Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics 2004; 20:1170 - 7; http://dx.doi.org/10.1093/bioinformatics/bth059; PMID: 14764558
- Price AL, Eskin E, Pevzner PA. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome Res 2004; 14:2245 - 52; http://dx.doi.org/10.1101/gr.2693004; PMID: 15520288