Abstract
We conducted a genome-wide DNA methylation analysis in CD19+ B-cells from chronic lymphocytic leukemia (CLL) patients and normal control samples using reduced representation bisulfite sequencing (RRBS). The methylation status of 1.8–2.3 million CpGs in the CLL genome was determined; about 45% of these CpGs were located in more than 23,000 CpG islands (CGIs). While global CpG methylation was similar between CLL and normal B-cells, 1764 gene promoters were identified as being differentially methylated in at least one CLL sample when compared with normal B-cell samples. Nineteen percent of the differentially methylated genes were involved in transcriptional regulation. Aberrant hypermethylation was found in all HOX gene clusters and a significant number of WNT signaling pathway genes. Hypomethylation occurred more frequently in the gene body including introns, exons, and 3′-UTRs in CLL. The NFATc1 P2 promoter and first intron was found to be hypomethylated and correlated with upregulation of both NFATc1 RNA and protein expression levels in CLL suggesting that an epigenetic mechanism is involved in the constitutive activation of NFAT activity in CLL cells. This comprehensive DNA methylation analysis will further our understanding of the epigenetic contribution to cellular dysfunction in CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the United States and accounts for approximately 11% of all hematologic neoplasms. Despite recent advances in the understanding of the underlying pathophysiology and in the treatment of this disease, CLL remains incurable. The clinical course of patients with CLL is heterogeneous; some patients experience rapid disease progression while others live for decades without requiring treatment.Citation1,Citation2 Since treatment of unselected early stage patients with alkylating agents at diagnosis offers no survival advantage over treatment at the time of disease progression, the current paradigm for newly diagnosed, early-stage CLL patients is to pursue a strategy of “watch and wait,” which defers treatment until the disease progresses.Citation2 However, this approach often leads to profound anxiety and emotional distress for patients with CLL.Citation1 Although the identification and validation of prognostic molecular markers (including surface markers ZAP70 and CD38, cytogenetic abnormalities, and IGHV mutational status) has resulted in refinements in the management of these patients,Citation3-Citation5 many of these biological assays are expensive, difficult to standardize and not widely available.Citation1 Thus, the discovery of biologically relevant factors that influence the heterogeneity and progression of CLL will not only promote our understanding of the disease process, but also will allow us to identify rational therapeutic approaches.
Epigenetic processes such as DNA methylation and histone modifications control packaging of DNA and have a direct impact on the function of the human genome. Numerous studies have demonstrated that aberrant epigenetic changes play an important role in tumor initiation and progression.Citation6 Aberrant DNA methylation changes in tumor suppressor genes such as DAPK1,Citation7 SFRP1,Citation8 ID4,Citation9 genes involved in apoptosis, cell cycle regulators p16 and p15Citation10 and prognostic markers ZAP70Citation11,Citation12 and TWIST2Citation13 have been identified in CLL patients. DNA methylation changes were also found to be associated with disease progression in the Eµ-TCL1 transgenic mouse model of CLL.Citation14 In an initial global screen for CpG island (CGI) methylation using a technique called restriction landmark genomic scanning (RLGS), DNA hypermethylation was found in CLL patients with a mean of 4.8% of CGIs affected.Citation15 Recently, several genome-wide DNA methylation studies using DNA methylation microarray analyses identified additional aberrantly methylated genes in CLL samples.Citation16-Citation18 Our studies, as well as many others, have also shown that some of these methylated genes had good correlation with the existing prognostic markers.Citation16-Citation19 In this study, we utilized the reduced representation bisulfite sequencing (RRBS) to identify aberrant DNA methylation changes at single-base resolution in CD19+ B-cells purified from the peripheral blood of CLL samples and normal control subjects. Differential methylation between CLL and normal B-cells was identified within sequences with different functional annotations including, but not limited to CGI, CGI shore and repetitive sequences. Examples of both hyper and hypomethylation were identified in the 5′ end regulatory region and gene body of many genes that could potentially function in the CLL pathogenesis and lead to aberrant gene expression.
Results
Genome-wide bisulfite sequencing in normal and CLL B-cell samples
To perform a genome-wide analysis of DNA methylation in CLL, we applied the RRBS approach to 11 CLL B-cell samples, 3 normal control samples including one each of normal CD19+, CD19+/ IgD+ naïve (NBC), CD19+/CD27+ memory B-cell sample (MBC) and three CLL cell lines (Mec-1, Mec-2 and Wac-3). We generated 20–30 million Illumina sequencing reads for each sample. Of these, 63% to 75% were successfully mapped to either strand of the human genome (hg18). The average sequencing depth per CpG was between 32X and 43X. We were able to consistently determine the methylation status of approximately 1.8–2.3 million CpGs (). Over 23,000 CGIs, which accounts for more than 80% of annotated CGIs in the genome, were examined. About 40% of CpGs analyzed were located in the 5′ end regulatory or coding sequences (Fig. S1A). The overall methylation levels of CpGs showed a similar bimodal distribution in all samples (Fig. S1B), which is consistent with previous reports.Citation20 Most of the CpGs located in the 5′ end regulatory regions were found to be unmethylated (methylation level < 0.25), while CpGs located in the gene body and intergenic regions were typically methylated (methylation level > 0.75). The overall distribution of CpGs, as well as methylated CpGs, in the repetitive sequences was also consistent between samples (Fig. S2A and B). shows a pair-wise comparison of the single-base resolution methylation data. The overall CpG methylation profiles among the three normal B-cell samples were highly correlated as the pair-wise Pearson correlation coefficients (R) ranged from 0.96–0.97. The correlations among CLL samples and between CLL and normal B-cells were also high (R ranged from 0.89 to 0.93). However, the correlation between the cell lines and primary normal and CLL B-cells was significantly lower as indicated by decreased R-values ranging from 0.72 to 0.82 ().
Table 1. Summary Statistics
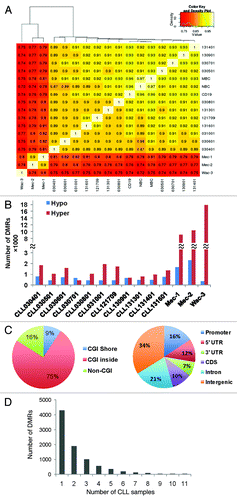
Figure 1. DNA methylation changes in CLL. (A) Pair-wise correlation coefficients matrix comparing DNA methylation between normal B-cells, CLL cell lines and primary CLL B-cells. A high similarity was observed among normal CD19+ B-cells (CD19), naïve (NBC) and memory B-cells (MBC), while CLL cell lines displayed significant differences when compared with the primary cells. (B) The number of DMRs identified in 3 CLL cell lines and 11 purified CLL B-cell samples when compared with three normal control samples. (C) Genome-wide distribution of 8703 DMRs that were identified in primary CLL samples. (D) Number of DMRs that are common among 11 CLL samples.
Identification of differentially methylated regions (DMRs)
To identify the DMRs between normal B-cells and CLL B-cells, we performed a genome-wide, unbiased DMR detection using a complete tiling of the human genome in 200 bp windows. Because adjacent CpGs are usually coordinately methylated,Citation21 we only performed analysis on windows with at least 5 sequenced CpGs. Using the criteria requiring an FDR q value < 0.01 and difference of average methylation level > 0.25, we identified several thousand differentially methylated windows in each primary CLL sample when compared with all three normal B-cell samples (). Figure S3A shows the volcano plots identifying DMRs between each CLL sample and the three normal control samples combined as a group. In the CLL samples, the number of hypomethylated DMRs was similar; however, the number of hypermethylated DMRs was quite variable (). In total, we identified 8703 DMRs that were hyper- or hypomethylated in at least one primary CLL sample as compared with all three normal control samples. We performed a permutation test to assess whether the identified DMRs were due to random variation between samples. We randomly re-assigned case-control status to the 11 cases and 3 controls, and then re-performed the genome-wide analysis and counted the number of DMRs observed. As shown in Figure S3C, after 100 permutations, none generated more DMRs than the original results with correct case-control status, suggesting that it was unlikely to obtain the original results by chance (p = 6.36 × 10−87). The majority of the 8703 DMRs (75%) were located within CGIs, and about 9% of the DMRs were located in the CGI shore regions. 28% of the DMRs were located in the promoter or 5′-UTR regions, while over half of the DMRs (55%) were located in the intergenic or intragenic regions (). Over half of the 8703 DMRs were differently methylated in only one CLL when compared with normal B-cell samples (). 353 DMRs were differentially methylated in more than 5 CLL samples and 52 DMRs presented in all 11 CLL samples.
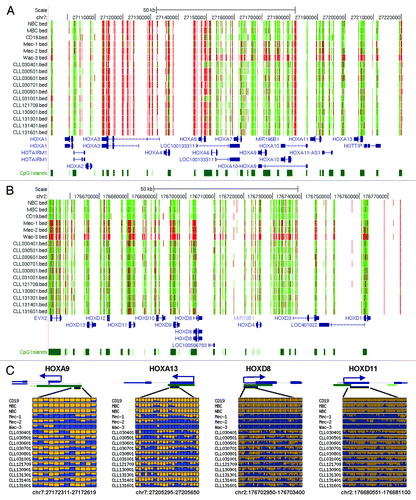
From the 8703 DMRs, we identified 1764 known genes that have DMRs located at the 5′ end regulatory regions (TSS ± 1000 bp). The functional annotation analysis generated using DAVID showed that about 18% (324 out of 1764) of these proteins regulate transcription, and 105 genes belong to the Homeobox protein family. There was significant enrichment in transcriptional regulators (p = 4.7 × 10−9, FDR-adjusted p value = 8.6 × 10−6) and Homeobox genes (p = 7.9 × 10−47, FDR-adjusted p value = 2.3 × 10−44). Aberrant methylation changes were observed in all four HOX gene clusters. As shown in , the CGIs associated with HOXA6 through HOXA13 were frequently hypermethylated in the CLL samples. Nearly all HOXD genes were found to be aberrantly hypermethylated in the CLL samples as well (). HOXA13, HOXD8 and HOXD11 were hypermethylated in all 11 CLL samples analyzed (). In the CLL cell lines, most of the HOX genes exhibited hypermethylation that was much denser and more uniform than was observed in primary CLL samples.
Figure 2. DNA hypermethylation of HOX genes. (A and B) UCSC genome browser screenshot illustrating the RRBS results in the HOXA and HOXD gene clusters. The tracks shown from the top to bottom in each figure are: the DNA methylation level at each CpG site derived from the bisulfite sequencing reads, RefSeq genes and annotated CpG islands in the UCSC genome browser. Red and green colors indicate methylated and unmethylated CpG sites, respectively. (C) Methylation profiles of individual HOX genes. In each panel, each row is the result of an individual patient sample. Each box represents a CpG site. Yellow, no methylation; blue, methylation. The proportion of yellow and blue in each box represents the methylation level. Only common CpGs shared by all samples are shown.
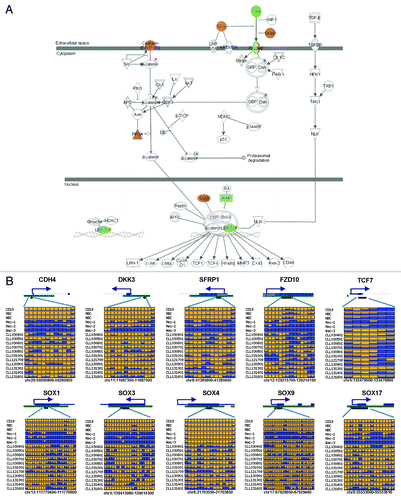
Based on the results from Ingenuity Pathway Analysis (IPA), the 1764 genes are enriched in several functional categories and canonical pathways (i.e., G-protein coupled receptor signaling, FDR-adjusted p-value = 4.57 × 10−5; cAMP-mediated signaling, FDR-adjusted p-value = 0.011; axon guidance signaling, FDR-adjusted p-value = 0.011; and WNT signaling, FDR-adjusted p-value = 0.014) (Tables S1 and S2). For instance, 29 of 174 WNT pathway genes were associated with DMRs located near the TSS in CLL samples. Notably, most of the hypermethylated genes in CLL were antagonists of WNT signaling such as members of DICKKOPF (DKK) and soluble frizzled protein (SFRP) families, as well as the SRY-like box (SOX) proteins (). The hypomethylated genes, on the other hand, involved the WNT ligands and TCF7 transcription factor ().
Figure 3. Aberrant DNA methylation changes in WNT pathway genes. (A) IPA analysis highlights DNA methylation alteration in WNT pathway genes. Hypermethylated genes are indicated by red and hypomethylated genes are highlighted by green. (B) Single-base CpG methylation patterns in WNT pathway genes in CLL. Each row is the result of an individual patient sample. Each box represents a CpG site. The color indicates the methylation level. Yellow, no methylation; blue, methylation. The proportion of yellow and blue in each box represents the methylation level. Only common CpGs shared by all samples are shown.
Differential methylation between normal memory and naïve B-cells
Using a similar approach as described above, we identified 1870 DMRs between NBC and MBC across the entire genome. Of those DMRs, 1706 were hypomethylated, while only 164 were hypermethylated in MBC as compared with NBC (Fig. S3B). Among the 152 genes associated with DMRs near the transcription start sites (TSS ± 1000 bp) (Table S3), 123 were hypomethylated and only 29 were hypermethylated in MBC when compared with NBC. IPA analysis showed that out of the 152 genes, 17 genes were involved in hematological system development and function, 10 genes were involved in hematopoiesis and 8 genes were involved in cell-mediated immune responses. Among them, Epstein-Barr virus induced 3 (EBI3), Interleukin-2 receptor α (IL2RA), programmed cell death 1 (PDCD1), tumor necrosis factor (ligand) superfamily, member 13B and 14 (TNFSF14 and TNFRSF13B) and myelin basic protein (MBP) play important roles in regulating the survival and proliferation of B and T-cells.
Identification of frequently hyper- and hypomethylated genes in CLL
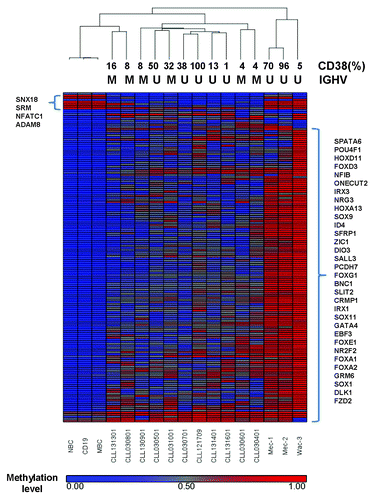
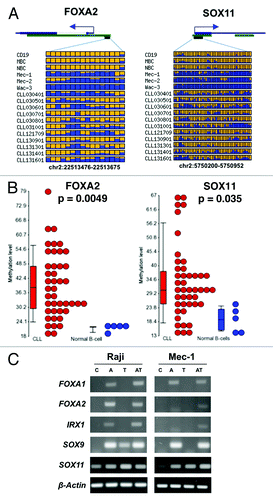
To identify the most frequently hyper- or hypomethylated genes in CLL, we performed Student's t-test analysis between CLL and normal B-cells using the average methylation value of each DMR. After the multiple testing adjustments, 533 out of 8703 DMRs survived the stringent statistical test (FDR q value < 0.05, methylation difference > 0.25). The detailed annotations of the 533 DMRs are listed in Table S4. Of these 533 DMRs, 400 DMRs were hypermethylated and 133 were hypomethylated. Furthermore, 158 DMRs were located at the 5′ end regulatory regions of 140 known genes. Cluster analysis using the average methylation values of the 158 DMRs clearly separated the CLL samples from three normal B-cell samples and cell lines (). However, the cluster analysis failed to further separate the CLL samples based on their unique clinical and molecular characteristics. Many frequently hypermethylated genes previously reported in CLL, as well as other hematological malignancies, such as: FOXD3,Citation14 FOXE1, FOXG1, IRX1,Citation15 ID4,Citation9,Citation14 SFRP1,Citation8 SLIT2,Citation22 BNC1, ADCY5, EBF3, NR2F2Citation18 and DIO3Citation23 were among the 140 genes (). Other frequently hypermethylated genes that have been previously reported in CLL, such as DAPK1, were among the initial list of 1764 genes, but failed to survive the statistical test. Several other SFRP proteins have been reported to be methylated in CLL;Citation8,Citation24 however, our study identified only SFRP1 as one of the most frequently hypermethylated genes in CLL. A subset of these genes was discovered to be differentially expressed between normal B-cells and CLL samples using an independent microarray study (unpublished results, McCarthy and Chiorazzi). The expression of SOX9 and SOX11 was also downregulated in CLL samples as compared with normal B-cells (Fig. S4).
Figure 4. Cluster analysis of 160 genes that associated with DMRs in the 5′ end regulatory regions (see text). The average methylation values of each DMR were subjected to hierarchical clustering with Pearson dissimilarity and average linkage method in Partek Genomics Suites. The IGHV mutation status and CD38 expression of each sample are given above the heatmap.
To confirm the DNA methylation results generated using RRBS, we performed bisulfite pyrosequencing to validate the methylation status of FOXA2 and SOX11 in an independent set of 43 CLL and 5 normal CD19+ B-cells samples (and S5). The results confirmed that FOXA2 and SOX11 were hypermethylated in a significant number of CLL patient samples (Student t-test, p = 0.0049 and 0.035, respectively). Treatment of two lymphoma cell lines with 5′-aza-2’-deoxycytidine (5′-Aza) and/or Trichostatin A (TSA) was able to successfully re-activate the expression of several hypermethylated genes in CLL including FOXA1, FOXA2, SOX9, SOX11 and IRX1 ().
Figure 5. Validation of DNA methylation results. (A) Methylation profiles of two frequently hyper-methylated genes generated by RRBS. Each box represents a CpG site. The color indicates the methylation level. Yellow, no methylation; blue, methylation. The proportion of the yellow and blue color indicates the levels of methylation. (B) Validation of DNA hypermethylation in the promoter and first exon of FOXA2 and SOX11 by bisulfite pyrosequencing, respectively. Each filled circle represents a CLL sample. The methylation values represent the average methylation levels of all CpG sites measured by pyrosequencing. Representative pyrosequencing results can be found in Figures S6 and S7. (C) Expression analysis of four selected genes in 2 lymphoma cell lines: total RNA isolated from treated (A, 5′-Aza; T, TSA; AT, 5′-Aza+TSA;) or untreated (C) cells was used to generate cDNA for RT-PCR analysis. β-actin was amplified for 20 cycles, while the other four genes were amplified for 32 cycles.
DNA hypomethylation in CLL
So far, most CLL methylation studies have been focused on promoter hypermethylation. RRBS analysis in CLL also identified genes frequently hypomethylated in CLL cells when compared with normal B-cells. Interestingly, 95 out of 133 (> 70%) hypomethylated DMRs were located in the gene body including exons, introns and 3′-UTRs (Table S4 Figure S6). Only 5 of the 133 hypomethylated DMRs were located in the promoter region. This was in contrast to the hypermethylated DMRs as over 40% of them were located in the 5′ end regulatory regions. Many genes that exhibited hypomethylation have a known or potential role as oncogenes; for instance, the oncogene TCL1A contained a hypomethylated DMR in the 3′-UTR (Fig. S6A). Hypomethylation was also identified within the gene body of BCR, LFNG, NOTCH1 (Fig. S6B), TCF7 (Fig. S6C), RASGRF1 and VAV2, as well as numerous other genes.
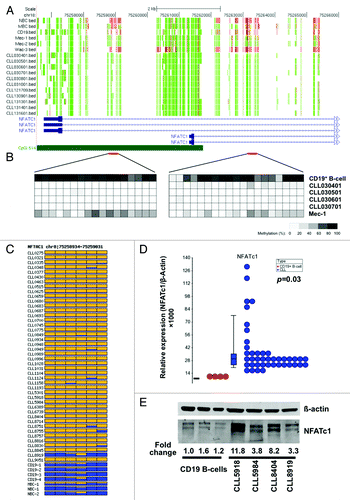
Among the hypomethylated genes, the promoter P2 and first intron regions of the transcription factor NFATc1 were found to be hypomethylated in all CLL patient samples examined by RRBS (). Conventional bisulfite genomic sequencing confirmed the differential methylation patterns in the 4 CLL samples and the Mec-1 CLL cell line that were analyzed using RRBS (). The bisulfite clone sequencing results were in complete agreement with the RRBS results. Pyrosequencing analysis was then conducted on the NFATc1 P2 promoter in 44 CLL samples, 4 normal CD19+ B-cell samples, 1 memory B-cell sample and 2 naïve B-cell samples used in the RRBS analysis. Figure S5C illustrates several representative pyrosequencing results. shows the summary of the pyrosequencing analysis for 6 CpGs in the NFATc1 P2 promoter in the CLL samples. Nearly all CLL samples contained significantly lower methylation levels in the promoter P2 (less than 10%), while the average methylation levels of NFATc1 in normal B-cells were more than 50%. The memory B-cell sample had the lowest methylation values when compared with naïve and CD19+ B-cells suggesting tissue specific differential methylation. We then analyzed the mRNA expression level of NFATc1 and found significant upregulation (Student t-test, p = 0.03) in CLL samples (). Western blot analysis of 4 CLL samples also indicated a 3 to 10-fold increase in NFATc1 protein expression ().
Figure 6. Hypomethylation and upregulation of NFATc1 expression in CLL. (A) RRBS results of NFATc1. The tracks show CpG sites that are covered by sequencing reads. The red and green colors indicate methylated and unmethylated CpGs, respectively. (B) Confirmation of DNA methylation in NFATc1 DMR using bisulfite clone sequencing. The methylation status of 36 CpGs from two regions (one in the promoter P2 and one in first intron) was determined from the bisulfite treated DNA of 4 CLL patients, one CLL cell line (Mec-1), and one normal CD19+ B-cell sample. Each row is the result of an individual clone. The same DNA samples used for RRBS sequencing were used in the confirmation study. (C) Quantitative pyrosequencing analysis of the NFATc1 P2 region in normal and CLL B-cells. Six CpGs in the promoter P2 region were analyzed by pyrosequencing. Each row is the result of an individual patient sample. Each box represents a CpG site. Yellow: no methylation; Blue: methylation. The proportion of yellow and blue in each box represents the methylation level. (D) Quantitative RT-PCR analysis of NFATc1 expression in normal and CLL B-cells samples. (E) Western blot analysis of NFATc1 expression in normal and CLL B-cells.
Discussion
In this study, we determined the methylation status of approximately 6–8% of the CpGs in the CLL genome using RRBS. These CpGs were highly enriched in CGI regions; over 23,000 CGIs were examined in normal and CLL B-cells. To our knowledge, this is the first sequencing-based methylation study in CLL. Previous studies using microarray or RLGS only analyzed up to approximately 28,000 CpG sites in the CLL genome. On average, only a few CpGs were examined in each CpG island or promoter region. At the single-CpG level, the global methylation pattern was similar between normal and CLL B-cells, as indicated by similar Pearson correlation coefficients among all primary B-cell samples. However, after scanning the genome using 200bp tiling windows, 8703 small DMRs were identified between CLL and normal B-cells, and about one third of the DMRs were located near the TSS. We have also identified tissue specific DMRs (tDMRs) between normal memory and naïve B-cells. The majority of the 1870 tDMRs (91%) were hypomethylated in memory B-cells as compared with naïve B-cells. This result appears to be in agreement with a recent genome-wide methylation study comparing germinal center (GC) B-cells with naïve B-cells, in which GC B-cells were found to be predominantly hypomethylated when compared with naïve B cells.Citation25 To determine whether the observed methylation differences could be due to variation in B-cell composition, or to normal variation between samples, we performed a permutation test and demonstrated that this was unlikely to be the case (p = 6.36 × 10−87).
We identified 1764 genes that were hyper- or hypomethylated in at least one CLL sample when compared with all three normal B-cell samples. The number of differentially methylated genes was quite variable between CLL samples suggesting heterogeneity among aberrant methylation changes in CLL. We further identified a group of 140 genes that were most commonly hypermethylated including several genes previously reported in CLL, as well as other hematological malignancies, such as, FOXD3,Citation14 FOXE1, FOXG1, IRX1,Citation15 ID4,Citation9,Citation14 SFRP1,Citation8 SLIT2,Citation22 BNC1, ADCY5, EBF3, NR2F2Citation18 and DIO3.Citation23 Interestingly, several large protein families such as HOX genes, FOX genes, and SOX genes were among the most frequently hypermethylated genes in CLL. The HOX gene family consists of four gene clusters located on chromosomes 2, 7, 12 and 17. We found that the HOXA, HOXC, and HOXD genes were the most affected by aberrant DNA methylation changes. Our study represents the first comprehensive interrogation of aberrant HOX gene methylation changes in CLL to date. Chen et al. showed that methylation-mediated silencing of FOXD3 was a key event early in leukemogenesis in both mouse and human CLL.Citation14 It was suggested that silencing of FOXD3 might lead to silencing of many of its target genes. In this study, we found that the methylation status of 20 FOX genes was altered in CLL including FOXD3. Several genes such as FOXA1 and FOXA2 are also potential downstream target genes of FOXD3.
Previous studies have determined that the WNT signaling pathway is activated in CLL.Citation26 The key transcription factor in the WNT pathway, LEF-1, has been shown to be overexpressed more than 28-fold in CLL B-cells.Citation27 We have found that most of the hypermethylated WNT pathway genes in CLL were antagonist proteins of WNT signaling such as DKK1, DKK3, and SFRP1. In addition, we found that 11 out of 20 SOX proteins were hypermethylated in CLL, including SOX9 which was identified as one of the most frequently hypermethylated genes in CLL. A recent study concluded that SOX9 inhibited β-catenin/TCF-dependent transcription and promoted β-catenin degradation by two separate mechanisms involving different domains of SOX9.Citation28 Epigenetic silencing of WNT signaling antagonists may disrupt the balanced inputs of these proteins and lead to the activation of this signaling network.
A significant number of genes were affected by hypomethylation in CLL, and most of the frequently hypomethylated DMRs were located in the gene body including coding sequences, introns, and 3′ UTRs. Intriguingly, the oncogene TCL1A was hypomethylated at the 3′ UTR. The impact of this aberrant epigenetic change on TCL1A expression is currently unclear and warrants further investigation. NOTCH1 is another important gene affected by hypomethylation in one of the exons. Hypomethylation of NOTCH1 in the 5′ regulatory region has previously been reported in mantle cell lymphoma;Citation29 however, we did not find similar changes in CLL. The significance and functional consequences of the hypomethylation in the gene body has not been well documented. However, these methylation changes may play an important role in regulating gene expression, particularly in a lineage or tissue specific manner. For example, the methylation status of conserved non-coding DNA elements in the Foxp3 gene can facilitate the heritable maintenance of the active state of the Foxp3 locus and determine the regulatory T-cell fate.Citation30
NFATc1 is a member of the nuclear factor of activated T-cells (NFAT) family of transcription factors and plays an epigenetic chromatin remodeling role in the transcriptional regulation of growth and survival genes including CD40L (CD40 ligand, also known as CD154) and BlyD (also known as BAFF).Citation31-Citation33 Recently it was reported that NFATc1 supports the proliferation and suppresses the activation-induced cell death of splenic B-cells upon B-cell receptor (BCR) stimulation.Citation34 Previous studies have shown that CLL contains constitutively activated NFAT transcription factor activity,Citation35 and immunohistochemistry staining has shown that NFATc1 is expressed in CLL.Citation36 Our data suggests that NFATc1 is overexpressed in CLL B-cells due to hypomethylation in both the promoter P2 and first intron regions. This hypomethylation occurred in 55 CLL samples studied (including both sequencing and validation studies) suggesting that epigenetic dysregulation of NFATc1 is a frequent event in CLL and may play an important role in the constitutive action of NFAT activity.
In summary, we used a single-base resolution bisulfite sequencing approach to characterize the DNA methylation map of purified B-cells from both normal control and CLL samples. We observed distinct patterns of DNA methylation in different functional elements across the genome. This study has uncovered not only several thousand novel cancer-specific DMRs, but also DMRs between subtypes of normal B-cells. Finally, hypomethylation of the transcription factor NFATc1 occurred in the majority of the CLL samples and causes significant upregulation of this gene. The digital methylation map generated in this study provides the precise genomic locations that undergo methylation changes. This map will be a highly valuable public resource for investigations aimed at understanding epigenetic regulation of the CLL genome.
Materials and Methods
Tissue samples and nucleic acid isolation
Blood samples were obtained from CLL patients at the Ellis Fischel Cancer Center (EFCC) in Columbia, MO (11 CLL samples), the GHSU Cancer Center in Augusta, GA (14 samples) and the North Shore-LIJ Health System in Manhasset, NY (29 samples) in compliance with the local Institutional Review Boards. Clinical and molecular characteristics of 11 CLL samples used for RRBS analysis are summarized in Table S5, and the CLL samples used for replication studies are summarized in Table S6. The CD19+ normal B-cell, CD19+/CD27+ memory B-cell, and CD19+/CD27- naïve B-cell samples used in the RRBS analysis were purchased from ALLCELLs Inc. Other normal CD19+ B-cell samples were purified from leuko-enriched blood samples purchased from a local blood bank. Mononuclear cell fractions (PBMCs) were isolated over a Ficoll-Hypaque density gradient. Freshly isolated B-cells were prepared by negative selection using RosetteSep B-cell isolation kit (StemCell Technologies). Naïve B-cell samples were enriched using a positive selection kit (StemCell Technologies). DNA was isolated using the QIAmp DNA Blood Mini kit (Qiagen). RNA was isolated using an RNeasy mini kit (Qiagen).
Cell culture and drug treatment
Three CLL cell lines were included in this study and were found to differ in their levels of CD38 expression, Wac-3 (4.7% CD38), Mec-1 (69.5% CD38), and Mec-2 (96.6% CD38).Citation17 CLL cell lines were maintained in RPMI 1640 media with 10% fetal bovine serum (FBS). A Burkitts lymphoma cell line, Raji, was also maintained in the media described above. For gene reactivation experiments, cells were cultured in the presence of vehicle (PBS) or 1.0 μM 5′-Aza with medium changed every 24 h. After 4 d, cells were either harvested, treated with TSA (1.0 μM) for an additional 12 h and then harvested, or treated solely with TSA for 12 h before being harvested. The total RNA was isolated as described above.
Reduced representation bisulfite sequencing
RRBS was performed according to a previously published protocolCitation20,Citation37 with minor modifications. For each sample, 1 μg genomic DNA was digested overnight using 40 units of MspI (New England Biolabs). The digested DNA was end-repaired and adenylated in a 50 μl reaction consisting of 10U of exo-Klenow fragments (Enzymatics), and 2 μl each of dGTP (1 mM), dATP (10 mM), and methylated dCTP (1 mM). The reaction was incubated at 30°C for 30 min followed by 37°C for an additional 30 min. The methylated Illumina adapters were ligated to the adenylated DNA fragments in a 20 μl reaction containing 2 μl concentrated T4 ligase (Enzymatics) at room temperature for 15 min. The ligation products were gel-selected for fragments with insertion sizes of 40 to 120 bp and 120 to 220 bp as previously suggested.Citation20,Citation37 Bisulfite treatment was conducted using the EZ DNA methylation kit (Zymo Research) according to manufacturer’s protocol. The final libraries were generated using 5 μl bisulfite-converted template in a 14-cycle PCR amplification using the PfuTurbo Cx Hotstart polymerase (Agilent Technologies). The libraries were sequenced using an Illumina Genome Analyzer IIx (Ilumina) with a read length of 52 or 76bp.
Real time RT-PCR
Total RNA was reverse transcribed in the presence of SuperScript II reverse transcriptase (Invitrogen). The cDNA was then analyzed using real time PCR performed using RTCitation2 SYBR green PCR master mix containing the appropriate reagents (SA Biosciences) in a LightCycler480 instrument (Roche). The primer sequences are listed in Table S7.
Immunoblot analysis
Normal and CLL B-cells were centrifuged and the cell pellets were re-suspended in lysis buffer. Forty μg of the protein lysate were separated on a 10% SDS-PAGE gel. The separated proteins were transferred to nitrocellulose membranes before being sequentially blocked for 1 h in LI-COR blocking buffer and incubated with primary antibody and secondary antibody with four 5 min washes in between. Membranes were scanned, and bands were quantified using the Odyssey infrared imaging system (LI-COR). The antibodies used were: anti-NFATc1 (7A6) (SC-7294), anti-β-Actin (C4) (sc-47778) from Santa Cruz Biotechnology and Goat anti-Mouse IgG IRDye800CW (827–0836) from LI-COR.
Bisulfite clone sequencing
Two regions in the promoter P2 and first intron of NFATc1 were analyzed using bisulfite clone sequencing. Primer sequences are listed in Table S7. The bisulfite conversion of genomic DNA was conducted as described above using 500ng of DNA for each sample. Amplified PCR products were sub-cloned using the TOPO TA Cloning Kit for sequencing (Invitrogen). Plasmid DNA of 16 insert-positive clones was isolated using the Qiagen Plasmid Miniprep kit and sequenced by Sanger sequencing.
Bisulfite pyrosequencing
The DMRs associated with FOXA2, SOX11, and NFATc1 were subjected to bisulfite pyrosequencing analysis. The bisulfite PCR and sequencing primers are listed in Table S7. The bisulfite treatment of genomic DNA was performed as described above. The pyrosequencing analysis was performed using PyroMark CpG assay reagents on a PyroMark Q24 instrument, according to manufacturer’s instructions (Qiagen). Program outputs were analyzed by the PyroMark Q24 software, and the percentage of methylated vs. unmethylated alleles was determined by calculating the ratio of relative peak heights.
Alignment of sequencing reads from bisulfite-treated DNA
The raw sequencing reads were cleaned using in-house scripts to trim sequencing adapters and low quality bases (Q < 67 in Illumina 1.5) in the 3′ end and ambiguous bases in both ends. To map the sequencing reads from RRBS, we extracted sequenceable regions from the human genome (hg18) that were within 100bp from the MspI sites at both ends. Each sequenceable region was indexed by converting all C’s and G’s to T’s and A’s, respectively, i.e., two different reference databases. Bowtie was used to map the cleaned reads to each of the two reference databases after converting all C’s to T’s. For each read, an in-house script computed the best of all alignments for the different loci using two different reference databases based on the number of mismatches after realigning the original read and reference sequences. The script also determined the methylation status of each cytosine residue by comparing the bisulfite-converted sequence to the reference sequence. Another in-house script piled reads for each cytosine in the reference genome and counted the numbers of reads that contained methylated and unmethylated cytosines, respectively. Finally the methylation of each CpG site was defined as the fraction of methylated reads to that of methylated and unmethylated reads combined. CpGs with < 5 reads were filtered out of further analyses. The correlation between genome-wide CpG methylation across two samples was calculated using the Pearson’s product-moment coefficient using an R script. The raw and analyzed sequencing data from this study has been submitted to NCBI Gene Expression Omnibus (http://ncbi.nlm.nih.gov/geo/) under accession number GSE32698.
Detection of DMRs
We used 200 bp non-overlapping windows to identify differentially methylated regions (DMRs). The windows containing fewer than 5 CpGs were filtered out of the further analysis. For each of these windows (> 5 CpGs), the number of methylated and unmethylated CpG observations was determined by summing the numbers of methylated and unmethylated CpGs in all reads that were mapped within each window, and a p value was assigned using Fisher exact tests. Once all p values were calculated, multiple-testing correction was performed separately for each window using the FDR q-value developed by Benjamini and Hochberg.Citation38 The DMRs between two samples (e.g., MBC vs. NBC) were identified with an FDR q value < 0.01 and a methylation difference > 0.25. To obtain the DMRs between two groups, i.e., CLL samples vs. normal B-cells, we first identified the DMRs between one CLL sample and each of the three normal B-cells samples by applying Fisher’s exact tests using the same stringent cutoff values in a pair wise fashion. Next, we selected only the common DMRs that were consistently identified in each CLL sample compared with all three normal B-cell samples. Finally, the DMRs discovered in each CLL sample when compared with the normal B-cell samples were merged to make up the DMRs between two groups.
Statistical analysis
Student's t-test analysis was used to identify differentially methylated genes with statistical significance between groups (i.e., CLL verses normal B-cells; IGHV mutated and unmutated CLL samples). The average methylation value of each window was used to perform the statistical analysis in R. The p-value was adjusted using FDR based on the method described above.Citation38 An FDR q value < 0.05 and a methylation difference > 0.25 were the cut off values used to identify the statistically significant differentially methylated genes.
| Abbreviations: | ||
| CGI | = | CpG island |
| CLL | = | chronic lymphocytic leukemia |
| RRBS | = | reduced representation bisulfite sequencing |
| DAVID | = | The Database for Annotation, Visualization and Integrated Discovery |
| RLGS | = | restriction landmark genomic scanning |
| DMR | = | differentially methylated regions |
| MBC | = | memory B-cells |
| NBC | = | naïve B-cells |
| PBMC | = | peripheral blood mononuclear cells |
Additional material
Download Zip (2 MB)Acknowledgments
We thank Drs. Nicholas Chiorazzi, Kanti R. Rai and Steven L. Allen for providing the CLL samples. We also thank Dr. Judith Giri and Ms. Sameera Qureshi of GHSU tumor bank for helping collect the CLL samples. We are grateful to Dr. Chiorazzi for sharing the unpublished microarray data with us. This work was supported in part by the National Institute of Health (Grants CA134304 and DA025779 to H.S.). H. S. is a Georgia Cancer Coalition Distinguished Cancer Scientist. J. C and H. T. were supported by a National Research Foundation of Korea Grant funded by the Korean Government (NRF-2009–352-D00275).
Conflict of interest
The authors declare no competing financial interests.
Related Research Data
Reference
- Shanafelt TD. Predicting clinical outcome in CLL: how and why. Hematology Am Soc Hematol Educ Program 2009; 2009:421 - 9; http://dx.doi.org/10.1182/asheducation-2009.1.421; PMID: 20008228
- Gribben JG. How I treat CLL up front. Blood 2010; 115:187 - 97; http://dx.doi.org/10.1182/blood-2009-08-207126; PMID: 19850738
- Butler T, Gribben JG. Biologic and clinical significance of molecular profiling in Chronic Lymphocytic Leukemia. Blood Rev 2010; 24:135 - 41; http://dx.doi.org/10.1016/j.blre.2010.03.004; PMID: 20398985
- Caligaris-Cappio F, Ghia P. Novel insights in chronic lymphocytic leukemia: are we getting closer to understanding the pathogenesis of the disease?. J Clin Oncol 2008; 26:4497 - 503; http://dx.doi.org/10.1200/JCO.2007.15.4393; PMID: 18662968
- Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood 2011; 117:1781 - 91; http://dx.doi.org/10.1182/blood-2010-07-155663; PMID: 21148333
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148 - 59; http://dx.doi.org/10.1056/NEJMra072067; PMID: 18337604
- Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007; 129:879 - 90; http://dx.doi.org/10.1016/j.cell.2007.03.043; PMID: 17540169
- Liu TH, Raval A, Chen SS, Matkovic JJ, Byrd JC, Plass C. CpG island methylation and expression of the secreted frizzled-related protein gene family in chronic lymphocytic leukemia. Cancer Res 2006; 66:653 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-3712; PMID: 16423993
- Chen SS, Claus R, Lucas DM, Yu L, Qian J, Ruppert AS, et al. Silencing of the inhibitor of DNA binding protein 4 (ID4) contributes to the pathogenesis of mouse and human CLL. Blood 2011; 117:862 - 71; http://dx.doi.org/10.1182/blood-2010-05-284638; PMID: 21098398
- Seeliger B, Wilop S, Osieka R, Galm O, Jost E. CpG island methylation patterns in chronic lymphocytic leukemia. Leuk Lymphoma 2009; 50:419 - 26; http://dx.doi.org/10.1080/10428190902756594; PMID: 19347729
- Chantepie SP, Vaur D, Grunau C, Salaün V, Briand M, Parienti JJ, et al. ZAP-70 intron1 DNA methylation status: determination by pyrosequencing in B chronic lymphocytic leukemia. Leuk Res 2010; 34:800 - 8; http://dx.doi.org/10.1016/j.leukres.2009.10.018; PMID: 19944462
- Corcoran M, Parker A, Orchard J, Davis Z, Wirtz M, Schmitz OJ, et al. ZAP-70 methylation status is associated with ZAP-70 expression status in chronic lymphocytic leukemia. Haematologica 2005; 90:1078 - 88; PMID: 16079107
- Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol 2005; 23:3877 - 85; http://dx.doi.org/10.1200/JCO.2005.02.196; PMID: 15809452
- Chen SS, Raval A, Johnson AJ, Hertlein E, Liu TH, Jin VX, et al. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2009; 106:13433 - 8; http://dx.doi.org/10.1073/pnas.0906455106; PMID: 19666576
- Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, et al. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res 2004; 64:2424 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-03-2870; PMID: 15059895
- Kanduri M, Cahill N, Göransson H, Enström C, Ryan F, Isaksson A, et al. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010; 115:296 - 305; http://dx.doi.org/10.1182/blood-2009-07-232868; PMID: 19897574
- Rahmatpanah FB, Carstens S, Hooshmand SI, Welsh EC, Sjahputera O, Taylor KH, et al. Large-scale analysis of DNA methylation in chronic lymphocytic leukemia. Epigenomics 2009; 1:39 - 61; http://dx.doi.org/10.2217/epi.09.10; PMID: 20495622
- Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, et al. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics 2010; 5:499 - 508; http://dx.doi.org/10.4161/epi.5.6.12179; PMID: 20484983
- Strathdee G, Sim A, Parker A, Oscier D, Brown R. Promoter hypermethylation silences expression of the HoxA4 gene and correlates with IgVh mutational status in CLL. Leukemia 2006; 20:1326 - 9; http://dx.doi.org/10.1038/sj.leu.2404254; PMID: 16688227
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:766 - 70; PMID: 18600261
- Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol 2010; 8:e1000533; http://dx.doi.org/10.1371/journal.pbio.1000533; PMID: 21085693
- Dunwell TL, Dickinson RE, Stankovic T, Dallol A, Weston V, Austen B, et al. Frequent epigenetic inactivation of the SLIT2 gene in chronic and acute lymphocytic leukemia. Epigenetics 2009; 4:265 - 9; PMID: 19550140
- Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, et al. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS One 2009; 4:e6986; http://dx.doi.org/10.1371/journal.pone.0006986; PMID: 19750229
- Chim CS, Pang R, Liang R. Epigenetic dysregulation of the Wnt signalling pathway in chronic lymphocytic leukaemia. J Clin Pathol 2008; 61:1214 - 9; http://dx.doi.org/10.1136/jcp.2008.060152; PMID: 18765431
- Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 2011; 118:3559 - 69; http://dx.doi.org/10.1182/blood-2011-06-357996; PMID: 21828137
- Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2004; 101:3118 - 23; http://dx.doi.org/10.1073/pnas.0308648100; PMID: 14973184
- Gutierrez A Jr., Tschumper RC, Wu X, Shanafelt TD, Eckel-Passow J, Huddleston PM 3rd, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood 2010; 116:2975 - 83; http://dx.doi.org/10.1182/blood-2010-02-269878; PMID: 20595513
- Topol L, Chen W, Song H, Day TF, Yang Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J Biol Chem 2009; 284:3323 - 33; http://dx.doi.org/10.1074/jbc.M808048200; PMID: 19047045
- Leshchenko VV, Kuo PY, Shaknovich R, Yang DT, Gellen T, Petrich A, et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood 2010; 116:1025 - 34; http://dx.doi.org/10.1182/blood-2009-12-257485; PMID: 20427703
- Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010; 463:808 - 12; http://dx.doi.org/10.1038/nature08750; PMID: 20072126
- Lapalombella R, Andritsos L, Liu Q, May SE, Browning R, Pham LV, et al. Lenalidomide treatment promotes CD154 expression on CLL cells and enhances production of antibodies by normal B cells through a PI3-kinase-dependent pathway. Blood 2010; 115:2619 - 29; http://dx.doi.org/10.1182/blood-2009-09-242438; PMID: 19965642
- Pham LV, Tamayo AT, Li C, Bueso-Ramos C, Ford RJ. An epigenetic chromatin remodeling role for NFATc1 in transcriptional regulation of growth and survival genes in diffuse large B-cell lymphomas. Blood 2010; 116:3899 - 906; http://dx.doi.org/10.1182/blood-2009-12-257378; PMID: 20664054
- Pham LV, Tamayo AT, Yoshimura LC, Lin-Lee YC, Ford RJ. Constitutive NF-kappaB and NFAT activation in aggressive B-cell lymphomas synergistically activates the CD154 gene and maintains lymphoma cell survival. Blood 2005; 106:3940 - 7; http://dx.doi.org/10.1182/blood-2005-03-1167; PMID: 16099873
- Bhattacharyya S, Deb J, Patra AK, Thuy Pham DA, Chen W, Vaeth M, et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin--NFAT signaling network. J Exp Med 2011; 208:823 - 39; http://dx.doi.org/10.1084/jem.20100945; PMID: 21464221
- Muzio M, Apollonio B, Scielzo C, Frenquelli M, Vandoni I, Boussiotis V, et al. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood 2008; 112:188 - 95; http://dx.doi.org/10.1182/blood-2007-09-111344; PMID: 18292287
- Marafioti T, Pozzobon M, Hansmann M-L, Ventura R, Pileri SA, Roberton H, et al. The NFATc1 transcription factor is widely expressed in white cells and translocates from the cytoplasm to the nucleus in a subset of human lymphomas. Br J Haematol 2005; 128:333 - 42; http://dx.doi.org/10.1111/j.1365-2141.2004.05313.x; PMID: 15667535
- Gu H, Bock C, Mikkelsen TS, Jäger N, Smith ZD, Tomazou E, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods 2010; 7:133 - 6; http://dx.doi.org/10.1038/nmeth.1414; PMID: 20062050
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 1995; 57:289 - 300