Abstract
The immunodeficiency, centromere instability and facial anomalies (ICF) syndrome is associated to mutations of the DNA methyl-transferase DNMT3B, resulting in a reduction of enzyme activity. Aberrant expression of immune system genes and hypomethylation of pericentromeric regions accompanied by chromosomal instability were determined as alterations driving the disease phenotype. However, so far only technologies capable to analyze single loci were applied to determine epigenetic alterations in ICF patients. In the current study, we performed whole-genome bisulphite sequencing to assess alteration in DNA methylation at base pair resolution. Genome-wide we detected a decrease of methylation level of 42%, with the most profound changes occurring in inactive heterochromatic regions, satellite repeats and transposons. Interestingly, transcriptional active loci and ribosomal RNA repeats escaped global hypomethylation. Despite a genome-wide loss of DNA methylation the epigenetic landscape and crucial regulatory structures were conserved. Remarkably, we revealed a mislocated activity of mutant DNMT3B to H3K4me1 loci resulting in hypermethylation of active promoters. Functionally, we could associate alterations in promoter methylation with the ICF syndrome immunodeficient phenotype by detecting changes in genes related to the B-cell receptor mediated maturation pathway.
Introduction
Alterations of the epigenetic landscape and single gene loci are associated to different types of diseases. Although cancer is the most frequently studied phenotype other diseases related to cardiovascular, neurological and autoimmune defects reveal variations regarding their epigenetic profiles.Citation1 The majority of observed alterations are affecting DNA methylation, a modification of cytosines introduced by DNA methyl-transferases (DNMTs). DNA hypermethylation in particular in gene promoter regions results in impaired transcription initiation and subsequent gene silencing.Citation2 While genome-wide the majority of cytosines in a CpG context are methylated, focal regions of low methylation level, often associated to CpG rich regions (CpG islands), secure the accessibility of the transcription machinery and hence facilitating gene activation.Citation3 Throughout cell division DNA methylation is maintained by DNMT1, whereas methyl-groups are introduced de novo by DNMT3A and DNMT3B.
Recombinant mouse models deficient for DNMT3B expression revealed profound developmental defects resulting lethality during embryo development or short after birth.Citation4 Interestingly, in humans homozygous mutations of DNMT3B, suggested to result in a reduced activity rather than a complete functional impairment, lead to the development of the immunodeficiency, centromere instability and facial anomalies (ICF) syndrome.Citation5,Citation6 The disease phenotype is characterized by immunodeficiency, due to decreased numbers of mature B cells causing agammaglobulinemia. Furthermore, chromosomal instability, mainly observed in juxtacentromeric regions of chromosome 1, 16 and to a lesser extent of chromosome 9, and facial anomalies are frequent disease features. Although the majority of genetic defects in ICF patients are related to DNMT3B (Type 1), genetic alteration in ZBTB24 were also associated to the disease phenotype (Type 2).Citation7,Citation8
From an epigenetic point of view DNA hypomethylation was observed at satellite repeats (SAT2/3) of chromosome 1, 9, 16 and 21.Citation9,Citation10 Of note, hypomethylation of SAT2 repeats on chromosome 1 and 16 and missegregation of both chromosomes was also observed after treatment with the DNMT inhibitor 5-Azacytidine.Citation11 RNAi mediated knockdown of DNMT3B in embryonic stem cells resulted in similar methylation changes as observed in the ICF patient. Here, whole-genome bisulphite sequencing (WGBS) revealed large hypomethylated regions in particular on the X chromosome and validated the DNA methylation loss in pericentric regions.Citation12 In addition to changes in DNA methylation, ICF patients reveal alterations in the expression of protein coding genesCitation13 and microRNAs.Citation14
Using WGBS of lymphoblastoid cell lines (LCLs) obtained from an ICF patient harboring mutated DNMT3B and one healthy control, we unraveled changes in DNA methylation at base-pair resolution and determined yet unknown alterations associated to the disease phenotype.
Results and Discussion
Previous studies identified epigenetic changes in ICF patients harboring DNMT3B mutations in terms of locus-specific DNA methylation and histone modifications.Citation13,Citation15 Here we focus on DNA methylation, the direct effect of altered DNMT3B function. To get an unbiased insight of changes in DNA methylation occurring genome-wide, we performed whole-genome bisulphite sequencing (WGBS) of lymphoblastoid cell lines (LCLs) obtained from a female ICF patient (caucasian, 1 y old) harboring mutated DNMT3B (A603T/STP807ins;Citation16,Citation17 ICF) and one healthy gender-matched control sample (CTRL; caucasian, 4 y old). All data are freely available at the Gene Expression Omnibus (GSE37578, www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37578). Overall, we generated 667 million reads for the CTRL and ICF samples from which 88.8 and 85.9% mapped uniquely to the reference genome (hg19), respectively. Genome-wide we obtained DNA methylation information for 93% and 90% of Cs and 94% and 93% of CGs with an average coverage of 9.9 and 9.5-fold for the CTRL and ICF sample, respectively. Comparing global CpG methylation level for both samples revealed profound decreased levels in the ICF patient. Here, the average methylation level of CpG sites dropped from 59.7% to 34.4% and in total we identified 8,955,695 (32%) significantly differentially methylated CpG sites (XCitation2 test, p < 0.05). The full set of WGBS data from CTRL and ICF is illustrated in (), using Circos.Citation18 In line with previously published reports using single loci approaches, we detected a decrease in methylation in centromeric satellite repeats of the chromosomes 1, 9 and 16. However, screening for differentially methylated site in an ICF patient at base-pair resolution for the first time revealed hypomethylated centromeric satellite regions on all other chromosome, even to larger extents (). In addition to the centromeres a global loss 41% (27–52%) of methylation was detected for all autosomes (). Most importantly, as previously suggested,Citation19 chromosome X showed more profound losses (63%) indicating a general failure in heterochromatic regions. Displaying average DNA methylation level along the chromosome determined additional hypomethylated regions outside of centromeric repeat-rich regions spanning the entire chromosome as displayed for chromosome 1 and X (). Overall, a loss of methylation was abundant along all genomic features, with losses detected in promoters, exons, introns, intragenic regions (). However the loss of DNA methylation was more intense in non-genic regions.
Figure 1. Whole genome bisulfite sequencing of a healthy (CTRL) and an ICF patient (ICF) sample. (A) Circos representation of genome-wide DNA methylation levels in the CTRL and ICF samples. Average levels for all the CGs in 297 10-Mbp-wide windows. Inner track indicates the magnitude of the difference between the CTRL and the ICF individual for each window (color scale and red line). Average methylation levels in all the regions are expressed as β values (0–1) and color-coded (blue). (B) DNA methylation level of centromeric regions of all 22 autosomes and the X chromosome displayed as ratio of methylated reads to unmethylated reads. (C) Genome-wide DNA methylation level of 22 autosomes and the X chromosome displayed as ratio of methylated reads to unmethylated reads. (D) DNA methylation profile of chromosome 1 for the CTRL (red) and the ICF sample (blue). (E) DNA methylation profile of chromosome X for the CTRL (red) and the ICF sample (blue). (F) DNA methylation level of genomic features displayed as ratio of methylated reads to unmethylated reads. (G) DNA methylation level of regulatory elements displayed as ratio of methylated reads to unmethylated reads. (H) DNA methylation profile of the promoter region (TSS +/− 2kb) of protein-coding genes for CpG rich (top) and poor (bottom) promoters of the CTRL (red) and ICF patient (blue) sample.
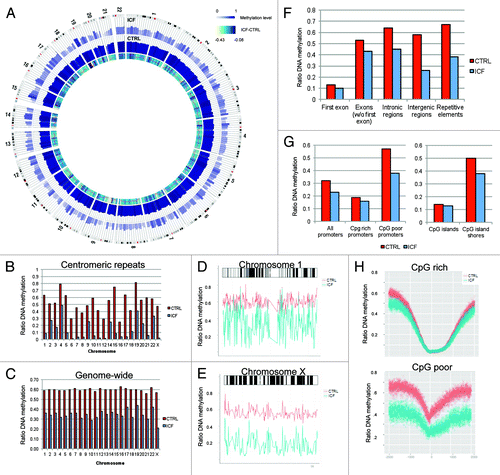
From a regulatory standpoint, DNA methylation decreased in CpG rich and poor promoters, however to a higher extend in the latter (). The same effect was observed for CpG islands, showing conserved hypomethylation at the CpG rich islands and an increased loss of DNA methylation at the island shores in the ICF patient. Interestingly, the shape of DNA methylation was conserved around the transcription start site (TSS) in CpG rich promoters, with changes preferentially occurring at the edges of the hypomethylated regions (). While genome-wide a drop of methylation from 60% to 34% was observed, proximate flanking regions of hypomethylated loci maintain a methylation level of around 50% suggesting the surrounding sites and shape of the hypomethylated region important to be preserved. In cancer cells CpG island shores were identified as crucial elements for gene regulation with high variable methylation level between normal and diseased tissue.Citation20,Citation21 In ICF patients aberrant methylation in CpG island shores might be responsible for previous reported changes in expression.Citation13 In addition to the conserved DNA methylation structure at promoters, CpG rich promoter containing genes were described as strongly expressed with a low evolutionary rate,Citation22 supporting a positive selection on epigenetic and genetic level. For CpG poor promoters, we observed a profound loss of DNA methylation at the TSS and the surrounding regions, however with conserved shape of the overall structure (). Comparing protein-coding and non-coding genes presented similar tendencies, with the non-coding RNA gene promoters showing general higher methylation levels (Fig. S1).
Regional enrichment of differential methylation
Displaying the methylation profile of the healthy control and ICF patient along the chromosomes, revealed a more profound drop of DNA methylation in the Giemsa-positive chromosomal regions, those representing loci of dense chromatin compaction (). Accordingly, the average methylation level significantly decreased in the ICF patient with Giemsa staining intensities (; Student’s t test; p < 0.01). As DNMT3B functions are closely related to chromatin structure, especially heterochromatin formation, we analyzed the DNA methylation profile of regions presenting distinct histone marks in more detail. Here, we took advantage of chromatin immunoprecipitation sequencing (ChIP-seq) experiments, enrolled in the ENCODE project processing a lymphoblastoid cell line of a healthy donor (GM12878), enabling us to determine positions occupied by distinct histone marks. Analyzing nine different histone marks and regions occupied by CTCF and H2A.Z for their average DNA methylation level on the concomitant positions, revealed a significant loss of methylation in the repressive Polycomb mark H3K27me3 loci, the repressive heterochromatin mark H3K9me3 lociCitation23,Citation24 and H4K20me1 loci, previously associated to repressedCitation25 and activeCitation23,Citation26 genes (; XCitation2 test; p < 0.01). All marks were previously associated to be enriched in the inactive X chromosome.Citation27 Furthermore, genes harboring the repressive histone mark H3K27me3 were previously associated with DNMT3B activity.Citation28 Also the CTCF and H2A.Z bound regions revealed a significant loss of DNA methylation (XCitation2 test; p < 0.05). While for CTCF a clear suppressive function was described, H2A.Z binding is rather associated to active gene promoters. However, in mice models an association to pericentric heterochromatin has been observed.Citation29 Most importantly, when we analyzed the autosomes separately, all aforementioned marks remained significantly altered, suggesting the regions to be affected genome-wide and not driven by the highly hypomethylated X chromosome. In contrary, histone marks clearly associated to transcriptional activity lost DNA methylation to a much lesser extent.Citation23 Particularly, the DNA methylation level of H3K79me2 and H3K36me3 enriched regions almost remained unchanged, by showing only 6% reductions and highly behaving against the genome-wide trend.
Figure 2. Distinct regions of aberrant methylation in the healthy (CTRL) and the ICF patient (ICF) sample. (A) DNA methylation level of genomic regions defined by Giemsa-staining. Displayed are all regions of one class (gneg: Giemsa-negative; gpos(25–100): Giemsa-positive (light-strong) as boxplot for the CTRL (red) and the ICF (blue) sample, with ** indicating significance (p < 0.01). (B) Average methylation level of genomic loci occupied by different histone marks, H2A.Z and CTCF. Displayed are the average DNA methylation levels in the analyzed regions, with significant differences indicated (** p < 0.01, * p < 0.05). (C) Relative reduction of average DNA methylation in different repetitive elements of the ICF sample compared with the CTRL. (D) Correlation of the DNA methylation status of neighboring CpGs in the CTRL (red) and the ICF (blue) sample. (E) Characterization of HMRs present in the CTRL sample in terms of DNA methylation level (Uscore) and CpG content. (F) Characterization of HMRs present in the ICF sample in terms of DNA methylation level (Uscore) and CpG content.
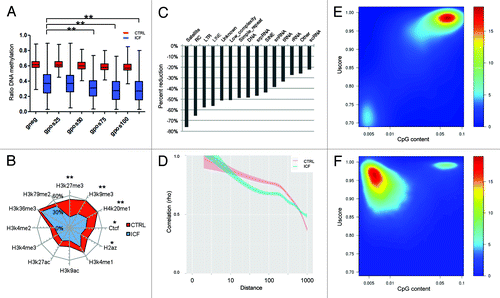
Furthermore, we aimed to analyze alterations occurring in repetitive elements in the ICF patient in more detail. Therefore, we extracted all sequencing reads with multiple mapping to the reference genome and analyzed them separately to extract DNA methylation changes in transposons and repeats. Strikingly, different repeat families appeared to be altered to variant extends, with satellite repeats revealing the highest reduction (-76%; ). Furthermore, transposons (RC, -66%; LINE, -58%, LTR, -57%) showed a very high reduction and RNA repeats (scRNA, -22%, rRNA, -27%, tRNA, -34%) the lowest reduction of DNA methylation, suggesting DNMT3B activity crucial for centromere stability as well as transposon repression ().
When we analyzed in particular the methylation status of neighboring CpG sites, we unexpectedly observed, regarding the massive changes observed genome-wide, that the DNMT3B mutant patients did not show significant less correlation between neighboring sites, suggesting the global hypomethylation to be due to a reduced activation, rather than a loss introduced by errors/failure of DNMT3B at random sites (). Therefore, we suggest that the here analyzed mutations are causing a reduction of activation of DNMT3B, while maintaining the enzyme’s processivity. This might be due to an impaired interaction with DNMT3L as previously shown for DNMT3B mutations in ICF patients.Citation30 Indeed, the A603T variant, known to be SAM binding deficient, also losses the ability to form homo-hetero-oligomers with DNMT3B and DNMT3L proteins in vitro and in vivo.Citation31,Citation32 As interaction of DNMT3B with DNMT3L highly increases the activity of the methyl-transferase,Citation33 we suggest that an impaired binding of DNMT3B to its activator is responsible for the global hypomethylation by paralleled conserved DNA methylation structures. Detecting an homogenously methylated genome in both samples suggests that the overall epigenomic landscape is maintained, consistent with the aforementioned maintenance of CpG rich promoter structures ().
Hypomethylated regions (HMRs)
As the ICF patient displayed a global loss of DNA methylation at high magnitude, we wondered if this affects the genome-wide structure of DNA methylation fingerprints such as hypomethylated regions (HMRs).Citation34,Citation35 HMRs are of special interest as they present loci of regulatory potential and were previously shown to alter their methylation level and shape following differentiation from hematopoietic stem/progenitor cells into myeloid or lymphoid lineages.Citation34 As an impaired maturation of B-cells is observed in ICF patients, deregulation of HMRs might contribute to the disease. Therefore, we assessed and characterized HMRs throughout the genome and compared these between both samples. We observed that the number of HMRs more than tripled in the ICF patients. While healthy cells harbored around 48,000 distinct regions of hypomethylation, the ICF patient reveals 164,000 HMRs. The changes are even more striking considering the size and CpG content of the HMRs in the patient samples as they cover 38.2% of the genome and harbor 39.3% of all CpGs. In contrary, HMRs in the healthy control sample represent only 3.1% of the genome and 9.9% of CpG sites. This is also reflected by the geometric mean sizes of 1.4 kb (IC 95%: 0.2–8.4 kb) vs. 5.0 kb (IC 95%: 0.8–30.9 kb) of the control and ICF sample, respectively.
To further characterize the distinct HMR populations in the healthy and ICF samples, we assessed HMR size, CpG content and scoreCitation34,Citation35 (determines the number of CpG sites in an HMR weighted inversely by their methylation status (more methylation, lower value). The Uscore presents a normalized score value (Uscore = score/CpG) to allow comparisons independent of HMR length. While the healthy cells revealed mainly CpG dense, highly hypomethylated loci and some CpG poorer regions with moderate hypomethylation, the ICF patient presents CpG poor regions with low to intermediate DNA methylation levels (). Although we observed a global hypomethylation of the genome displayed by large blocks of reduced DNA methylation, a subpopulation of HMRs kept their size and composition probably enabling ubiquitous expression by structural conservation of gene promoters crucial for cell survival. These conserved regions displayed a high CpG content and low level of DNA methylation, features frequently observed at CpG rich promoters, previously identified to maintain their DNA methylation structure.
Differentially methylated regions (DMRs)
In order to identify specific loci differentially methylated between the control and ICF sample, we screened the methylomes for regions consistently changing between both samples. DMRs were defined as regions of at least five consistently differentially methylated CpG sites between the control and ICF sample. In total, we assigned 315,813 DMRs present in all chromosomes. Consistent with before reported genome-wide changes, the DMR represented almost exclusively hypomethylation (296,964 out of 315,813) in the ICF patient (). Interestingly, despite the global loss of methylation distinct regions displayed a gain of methylation level in the ICF patient. In particular, we found 18849 (6.0%) DMRs representing a gain of methylation in the ICF patient, displaying a distinct genomic distribution compared with their hypomethylated counterparts. The majority of hypo-DMRs (57.8%) mapped to intergenic regions, 37.7% were overlapping gene bodies (intragenic) and 4.5% were located in gene promoters. In contrast, the majority of hyper-DMRs were associated to genic regions with 66.6% and 11.8% mapping to gene bodies and promoter regions, respectively. Analyzing the chromosomal distribution of DMRs revealed an increased abundance on the X chromosome than on the autosomes with 55% and 41% of the chromosomes covered by the DMRs, respectively.
Figure 3. Differentially methylated regions (DMRs) in the healthy (CTRL) and the ICF patient (ICF) sample. (A) Distribution of DMR observed between the samples in terms of DNA methylation level and genomic location. (B) DNA methylation profiles of the promoter regions of the germline-specific genes TDRD1 (left) and TDRD9 (right) in the CTRL (green) and the ICF (blue) sample. DMRs (red bars) and CpG islands (black bars) are indicated. (C) Enrichment ratios (positive value: overrepresentation; negative values: underrepresentation) of hyper-DMRs in different histone marks, H2A.Z and CTCF. (D) Relative position counts of hyper-DMRs in H3K4me1 marked promoters. Displayed are relative counts for H3K4me1 occupancy and hyper-DMRs in a 10 bp window. (E) DNA methylation profile of promoter regions (TSS +/− 2kb) overlapping with hyper-DMRs of the CTRL (red) and the ICF (blue) sample. (F) DNA methylation profiles of the promoter regions of the B-cell maturation associated genes VAV1 (left), NCK1 (middle) and GBR2 (right) in the CTRL (green) and the ICF (blue) sample. DMRs (red bars) and CpG islands (black bars) are indicated. (G) DNA methylation profiles of the promoter regions of the BTK activator SYK (left) and the BTK repressor SH3BP5 (right) in the CTRL (green) and the ICF (blue) sample. DMRs (red bars) and CpG islands (black bars) are indicated.
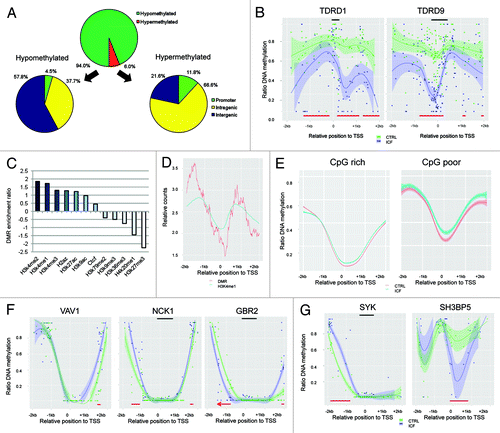
Although genomic hypomethylation appeared throughout the genome, the loss of methylation at CpG rich gene promoters did not present diffuse pattern but appeared rather organized ensuring expression of genes crucial for survival. In line, we detected tissue-specific genes highly methylated in the healthy control that revealed a de novo established HMR in their promoters. Here in particular testis-cancer specific genes of the TUDOR family (TDRD1 and TDRD9) presented sharply structured de novo HMRs in their promoter () accompanied by re-expression of the testis-specific genes in B-cells of ICF patients, as previously detected.Citation13
In the here analyzed disease context hypermethylated regions represent a special event as they behave against the global loss of DNA methylation, but also against the expectations, as ICF patients harbor mutations in DNMT3B, repressing its activity. DNMT3B binding to H3K4 was previously described to be impaired by DNMT3L upon mono-, bi- or tri-methylation of the H3K4 residue. In ICF patient, we detected an enrichment of hypermethylated DMRs in particular at methylated H3K4 marked loci, suggesting the residual activity of DNMT3B to be misguided to those regions ().Citation36 Here, mislocated DNMT3B activity might be mediated by an impaired interaction with DNMT3L as previously determined for different DNMT3B mutants in ICF patients.Citation30 DNMT3L interacts with unmethylated H3K4 tails, whereas H3K4 methylation blocks the binding and subsequent DNA methylation of the marked regions by DNMT3B.Citation33,Citation37 Consistently, we detected an enrichment of hypermethylated DMRs at sites previously inhibited by DNMT3L binding such as methylated H3K4 (). As especially H3K4me1 marks the boundaries flanking the TSSCitation23 ( andS2), an mislocated activity of DNMT3B in the flanking region could be suspected. Indeed, displaying promoter sites harboring hypermethylated DMRs revealed an increase of methylation in the sharply dropping boundaries, resulting in more narrow HMRs of CpG prich and poor promoters (). In addition, hyper-DMRs reveal a promoter distribution highly similar to the positioning of H3K4me1 (). Here, chromatin immunoprecipitation bisulphite sequencing (ChIP-BS-seqCitation38) of H3K4me1 of healthy and ICF samples could clearly clarify if these regions gain methylation in the patient.
Analyzing genes harboring hyper-DMRs in their respective promoter regions by Gene Ontology (GO) analysis (Biological process, level 5) revealed an enrichment of basic cellular mechanisms, such as RNA metabolism, regulation of transcription, cellular biosynthesis, nucleobase, nucleoside, nucleotide and nucleic acid metabolism, macromolecule biosynthesis and transferase activity (FDR, p < 0.05).
Therefore we propose a model in which for certain constitutively active housekeeping genes the promoter structure is maintained to ensure transcription. However, these genes present slightly narrowed gene promoter HMRs in ICF patients, suggesting that the responsible mechanism is failing to entirely restore the original status of the healthy donor (). We suggest that mislocated DNMT3B activity is hypermethylating sharp promoter boundaries, which are marked and protected by H3K4me1 in the original state (). In wild-type cells binding of DNMT3B to methylated H3K4 is impaired by DNMT3L resulting in hypomethylated H3K4 marked regions and wider promoter HMRs.
From a disease point of view, genes gaining methylation are associated to B-cell maturation, a function impaired in ICF patients causing agammaglobulinemia and severe immunological defects. Although based on different genetic alteration, ICF patients share phenotypical overlap with patients suffering from congenital agammaglobulinemia,Citation39 which also present an early onset of recurrent infections. This X chromosome-linked linked disease is caused by mutation of the Bruton’s tyrosine kinase (BTK), resulting in an impaired B-cells maturation and immune system failure. BTK is involved in the signal transduction activated by the B-cell receptor (BCR), which involves additional molecules such as SYK and the B-cell linker (BLNK), a scaffold protein that binds BTK, PLCγ2, GRB2, VAV1 and NCK1.Citation39,Citation40 Considering similarities in the disease phenotype of congenital agammaglobulinemia and ICF, it is tempting to speculate that the BCR pathway is also altered in ICF, although not by genetic but epigenetic alterations. Consistently, we found hypermethylated DMRs in the promoter region of GRB2, VAV1 and NCK1 creating smaller HMRs overlapping the TSS (). In line, we found further genes of the BCR pathway epigenetically altered. In particular, the BTK activator SYK revealed a hypermethylated DMR and the BTK repressor SH3BP5Citation41 a hypomethylated DMR in their promoter regions (). It is of note that the hypomethylation of the SH3BP5 promoter was not diffuse, but rather creating a sharply formed HMR likely to favor genes expression through its transcription promoting structure. Most importantly, the expression of SYK and SH3BP5 were shown to be down- and upregulated in ICF patients, respectively, consistent with their promoter methylation profiles.Citation13 Interestingly, ZBTB24, the second gene genetically altered in ICF Type 2 patients harbored a hypermethylated DMR in its promoter. Although not mutated, inactivating hypermethylation of ZBTB24 might contribute to the Type 1 disease phenotype as apparent in ICF Type 2 patients.
Conclusion
Although ICF patients show a massive loss of DNA methylation throughout all genomic regions and genetic features, some structures are maintained in their original state, suggesting a selective pressure to keep those features stable to ensure cell survival. Despite a global loss of DNA methylation in DNMT3B mutant cells, the shape of genetic features, such as promoters or CpG islands is conserved. Approaching genetic structures by the analysis of hypomethylated region supports the idea of structural addiction to ensure survival as a subpopulation of small CpG rich loci with low levels of methylation was conserved in the ICF patient, against the global trend of wide hypomethylated regions. Furthermore, DNMT3B mutant cells do not show a decrease of correlation between neighboring CpG sites favoring the idea that although flattened, the global DNA methylation landscape is maintained. Unexpectedly, distinct hypermethylated loci were observed and associated to H3K4 occupied regions, suggesting a mislocated activity of DNMT3B in ICF patients due to the lack of control by DNMT3L. As DNMT3L binding to DNMT3B also highly increases the activity of the methyl-transferase, the impaired interaction reduces both, activity and specificity of the enzyme, with effects visible in the ICF patient’s DNA methylome.
Functionally, ICF patients present hypermethylated genes involved in B-cell maturation and previously associated to congenital agammaglobulinemia. Induction of these genes by DNMT inhibitors, such as 5-Azacytidine, might serve as possible treatment strategy for ICF patients mutant for DNMT3B.
Materials and Methods
Sample preparation
Normal B cells of an healthy donor (female, 4 y) were immortalized by Epstein-Barr-Virus (EBV) applying previous published protocol.Citation42 Lymphoblastoid cell line generated from an ICF patient (GM08714, female, 1 y) was obtained from the Coriell Cell Repositories. DNA was extracted using Phenol:Chloroform:Isoamylalcohol (Sigma).
Whole-genome bisulphite sequencing
For normal BS-seq library constructing, 10μg genomic DNA was fragmented using a Covaris sonication system (Covaris S2). Following fragmentation, libraries were constructed using the Illumina Paired-End protocol consisting of end repair, < A > base addition and methylated-adaptor ligation. Ligated DNA was bisulfite converted using the EZ DNA Methylation-Gold kit (ZYMO). Different insert size were excised from the same lane of a 2% TAE agarose gel. Products were purified by using QIAquick Gel Extraction kit (Qiagen) and amplified by PCR. PCR was performed in a final reaction volume of 50μl consisting of 20μl purified DNA, 4μl 2.5 mM dNTP, 5 μl 10X buffer, 0.5 μl JumpStart™ Taq DNA polymerase, 2μl 10uM PCR primers and 37.5 μl water and the following thermal cycling program: 94°C 30 sec, 10 cycles of 94°C 30 sec, 60°C 30 sec, 72°C 30 sec then prolong with 1 min at 72°C. Sequencing was performed using the HighSeq2000 (Illumina).
Data processing
We perform the sequences alignment and methylation calling with version 2.43 of BSMAP software.Citation43 All genomic information, unless otherwise expressly stated, was obtained using CellBase. SAM/BAM and BED files handling was done using SAMtoolsCitation44 and bedtools.Citation45 Statistical analysis and graphic representation was performed with R (http://www.R-project.org) and libraries multicore and ggplot2. We defined the promoter region as 2 kb flanking the transcription start site (TSS). TSS was considered to be the upstream-most base of all the transcripts of the gene. When depicting methylation profiles, only CpG dinucleotide sites with coverage above or equal to 4 were considered.
Distance correlation
We assessed distance correlation of CpG in close proximity gathering information about methylation and relative distance up to 1000 bases away from 2000 randomly selected CpG sites and correlating pairwise methylation at single CpG sites for each relative distance.
Hypomethylated regions
The hypo-methylated regions (HMR) were identified via a hidden Markov model segmentation algorithm as previously described by Molaro et al.Citation35
Differentially methylated regions
Differentially methylated regions (DMR) were identified by considering all homozygous CpG dinucleotides, and searching for maximal length sequences of consecutive CpG’s with a consistent direction of methylation change between the two samples for all CpG’s in the region, and where the first and last CpG dinucleotides of a regions that contains minimum 5 CpGs showed a χ2 statistic for differential methylation of > 3.84, corresponding to a p value of < 0.05 for both residues.
Additional material
Download Zip (209.8 KB)Acknowledgments
The research leading to these results has received funding from the European Research Council (ERC) grant EPINORC under the agreement n° 268626, the MICINN Project – SAF2011–22803, the Cellex Foundation, the European Community's Seventh Framework Programme (FP7/2007–2013) by the grants PITN-GA-2009–238242 – DisChrom and HEALTH-F5–2011–282510 – BLUEPRINT and the Health and Science Departments of the Generalitat de Catalunya. ME is an ICREA Research Professor.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
H.H. and M.E. conceived and designed the experiments. All authors analyzed the data. H.H. and M.E. wrote the manuscript.
Related Research Data
References
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 2010; 28:1057 - 68; http://dx.doi.org/10.1038/nbt.1685; PMID: 20944598
- Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358:1148 - 59; http://dx.doi.org/10.1056/NEJMra072067; PMID: 18337604
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 66; http://dx.doi.org/10.1038/ng1990; PMID: 17334365
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915 - 26; http://dx.doi.org/10.1016/0092-8674(92)90611-F; PMID: 1606615
- Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999; 402:187 - 91; http://dx.doi.org/10.1038/46214; PMID: 10647011
- Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A 1999; 96:14412 - 7; http://dx.doi.org/10.1073/pnas.96.25.14412; PMID: 10588719
- Chouery E, Abou-Ghoch J, Corbani S, El Ali N, Korban R, Salem N, et al. A novel deletion in ZBTB24 in a Lebanese family with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin Genet 2011; http://dx.doi.org/10.1111/j.1399-0004.2011.01783.x; PMID: 21906047
- de Greef JC, Wang J, Balog J, den Dunnen JT, Frants RR, Straasheijm KR, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet 2011; 88:796 - 804; http://dx.doi.org/10.1016/j.ajhg.2011.04.018; PMID: 21596365
- Brun M-E, Lana E, Rivals I, Lefranc G, Sarda P, Claustres M, et al. Heterochromatic genes undergo epigenetic changes and escape silencing in immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome. PLoS One 2011; 6:e19464; http://dx.doi.org/10.1371/journal.pone.0019464; PMID: 21559330
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999; 99:247 - 57; http://dx.doi.org/10.1016/S0092-8674(00)81656-6; PMID: 10555141
- Prada D, González R, Sánchez L, Castro C, Fabián E, Herrera LA. Satellite 2 demethylation induced by 5-azacytidine is associated with missegregation of chromosomes 1 and 16 in human somatic cells. Mutat Res 2012; 729:100 - 5; http://dx.doi.org/10.1016/j.mrfmmm.2011.10.007; PMID: 22032830
- Martins-Taylor K, Schroeder DI, Lasalle JM, Lalande M, Xu R-H. Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics: Official Journal of the DNA Methylation Society 2012; 7.
- Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet 2008; 17:690 - 709; http://dx.doi.org/10.1093/hmg/ddm341; PMID: 18029387
- Gatto S, Della Ragione F, Cimmino A, Strazzullo M, Fabbri M, Mutarelli M, et al. Epigenetic alteration of microRNAs in DNMT3B-mutated patients of ICF syndrome. Epigenetics 2010; 5:427 - 43; http://dx.doi.org/10.4161/epi.5.5.11999; PMID: 20448464
- Matarazzo MR, De Bonis ML, Gregory RI, Vacca M, Hansen RS, Mercadante G, et al. Allelic inactivation of the pseudoautosomal gene SYBL1 is controlled by epigenetic mechanisms common to the X and Y chromosomes. Hum Mol Genet 2002; 11:3191 - 8; http://dx.doi.org/10.1093/hmg/11.25.3191; PMID: 12444103
- Wijmenga C, Hansen RS, Gimelli G, Björck EJ, Davies EG, Valentine D, et al. Genetic variation in ICF syndrome: evidence for genetic heterogeneity. Hum Mutat 2000; 16:509 - 17; http://dx.doi.org/10.1002/1098-1004(200012)16:6<509::AID-HUMU8>3.0.CO;2-V; PMID: 11102980
- Jiang YL, Rigolet M, Bourc’his D, Nigon F, Bokesoy I, Fryns JP, et al. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum Mutat 2005; 25:56 - 63; http://dx.doi.org/10.1002/humu.20113; PMID: 15580563
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009; 19:1639 - 45; http://dx.doi.org/10.1101/gr.092759.109; PMID: 19541911
- Hansen RS, Stöger R, Wijmenga C, Stanek AM, Canfield TK, Luo P, et al. Escape from gene silencing in ICF syndrome: evidence for advanced replication time as a major determinant. Hum Mol Genet 2000; 9:2575 - 87; http://dx.doi.org/10.1093/hmg/9.18.2575; PMID: 11063717
- Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 2009; 41:1350 - 3; http://dx.doi.org/10.1038/ng.471; PMID: 19881528
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Tang CSM, Epstein RJ. A structural split in the human genome. PLoS One 2007; 2:e603; http://dx.doi.org/10.1371/journal.pone.0000603; PMID: 17622348
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001; 410:120 - 4; http://dx.doi.org/10.1038/35065138; PMID: 11242054
- Karachentsev D, Sarma K, Reinberg D, Steward R. PR-Set7-dependent methylation of histone H4 Lys 20 functions in repression of gene expression and is essential for mitosis. Genes Dev 2005; 19:431 - 5; http://dx.doi.org/10.1101/gad.1263005; PMID: 15681608
- Vakoc CR, Sachdeva MM, Wang H, Blobel GA. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol 2006; 26:9185 - 95; http://dx.doi.org/10.1128/MCB.01529-06; PMID: 17030614
- Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev 2006; 20:1848 - 67; http://dx.doi.org/10.1101/gad.1422906; PMID: 16847345
- Choi SH, Heo K, Byun H-M, An W, Lu W, Yang AS. Identification of preferential target sites for human DNA methyltransferases. Nucleic Acids Res 2011; 39:104 - 18; http://dx.doi.org/10.1093/nar/gkq774; PMID: 20841325
- Rangasamy D, Berven L, Ridgway P, Tremethick DJ. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J 2003; 22:1599 - 607; http://dx.doi.org/10.1093/emboj/cdg160; PMID: 12660166
- Xie Z-H, Huang Y-N, Chen Z-X, Riggs AD, Ding J-P, Gowher H, et al. Mutations in DNA methyltransferase DNMT3B in ICF syndrome affect its regulation by DNMT3L. Hum Mol Genet 2006; 15:1375 - 85; http://dx.doi.org/10.1093/hmg/ddl059; PMID: 16543361
- Moarefi AH, Chédin F. ICF syndrome mutations cause a broad spectrum of biochemical defects in DNMT3B-mediated de novo DNA methylation. J Mol Biol 2011; 409:758 - 72; http://dx.doi.org/10.1016/j.jmb.2011.04.050; PMID: 21549127
- Ueda Y, Okano M, Williams C, Chen T, Georgopoulos K, Li E. Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development 2006; 133:1183 - 92; http://dx.doi.org/10.1242/dev.02293; PMID: 16501171
- Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007; 448:714 - 7; http://dx.doi.org/10.1038/nature05987; PMID: 17687327
- Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell 2011; 44:17 - 28; http://dx.doi.org/10.1016/j.molcel.2011.08.026; PMID: 21924933
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 2011; 146:1029 - 41; http://dx.doi.org/10.1016/j.cell.2011.08.016; PMID: 21925323
- Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res 2010; 38:4246 - 53; http://dx.doi.org/10.1093/nar/gkq147; PMID: 20223770
- Hu J-L, Zhou BO, Zhang R-R, Zhang K-L, Zhou J-Q, Xu G-L. The N-terminus of histone H3 is required for de novo DNA methylation in chromatin. Proc Natl Acad Sci U S A 2009; 106:22187 - 92; http://dx.doi.org/10.1073/pnas.0905767106; PMID: 20018712
- Brinkman AB, Gu H, Bartels SJ, Zhang Y, Matarese F, Simmer F, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 2012; http://dx.doi.org/10.1101/gr.133728.111; PMID: 22466170
- Conley ME. Genes required for B cell development. J Clin Invest 2003; 112:1636 - 8; PMID: 14660738
- Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev 2009; 228:132 - 48; http://dx.doi.org/10.1111/j.1600-065X.2008.00748.x; PMID: 19290925
- Yamadori T, Baba Y, Matsushita M, Hashimoto S, Kurosaki M, Kurosaki T, et al. Bruton’s tyrosine kinase activity is negatively regulated by Sab, the Btk-SH3 domain-binding protein. Proc Natl Acad Sci U S A 1999; 96:6341 - 6; http://dx.doi.org/10.1073/pnas.96.11.6341; PMID: 10339589
- Miller G, Lipman M. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc Natl Acad Sci U S A 1973; 70:190 - 4; http://dx.doi.org/10.1073/pnas.70.1.190; PMID: 4346033
- Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 2009; 10:232; http://dx.doi.org/10.1186/1471-2105-10-232; PMID: 19635165
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078 - 9; http://dx.doi.org/10.1093/bioinformatics/btp352; PMID: 19505943
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841 - 2; http://dx.doi.org/10.1093/bioinformatics/btq033; PMID: 20110278