Abstract
Histone deacetylase complexes (HDACs) are powerful regulators of the epigenome. It is now clear that a subset of HDACs also regulate the stability of the genome itself, but not primarily through transcription. Instead, these key HDACs control genome stability more directly by stabilizing enzymes important for DNA mutagenesis and repair, or by modifying histones at sites of DNA damage. Surprisingly, certain HDACs in budding yeast and human cells accelerate the pace of genetic expansions in trinucleotide repeats, the type of mutation that causes Huntington disease. In other words, HDACs promote mutagenesis in some settings. At double-strand breaks, however, the same HDACs in budding yeast help stabilize the genome by facilitating homology-dependent repair. Double-strand breaks can also be repaired without the requirement for homology, and two specific human HDACs are now known to promote this event. These new findings highlight certain HDACs as caretakers of genome stability, and also underscore the potential medical complexities in using HDAC inhibitors for treatment of disease.
Trinucleotide Repeat Expansions
Repeats of three-base elements such as CAG are common in the human genome. Most repeats are genetically stable and benign, but some trinucleotide repeats (TNRs) expand—gain length—to cause heritable human neurological diseases such as Huntington’s disease.Citation1-Citation3 These relentless mutations can occur at exceptionally high frequencies, approaching 100% in some cases.Citation4 This remarkable finding means that TNR expansions are the norm, not the exception, in affected families. Furthermore, high expansion frequencies led to the inference that cellular proteins actually promote (favor) expansions,Citation5 despite the detrimental effects of these mutations. These proteins generally protect genome stability, but it is thought that the TNR DNA ‘corrupts’ their normal activities, leading inadvertently to expansions.Citation1,Citation3,Citation6 Transgenic mice with long TNRs also display extremely high expansion rates, which allowed the identification of several DNA repair proteins as promoting factors, based on the fact there are fewer expansions in mice lacking these repair proteins.Citation7-Citation14 These findings are consistent with misrepair of DNA as a source of TNR expansions.
We recently reported that specific HDACs are novel promoting factors for TNR expansions in budding yeast and human cells.Citation15 In other words, HDACs are causal in the mutational process of going from short to expanded repeats. Thus, HDACs join DNA repair proteins as promoting factors for TNR expansions. It is important to distinguish this finding from what is already known about differences in chromatin structure for normal-length and expanded TNRs, where the latter tend be in a more heterochromatic environment.Citation16 Differences in chromatin structure are especially important for diseases like fragile X syndrome and Friedreich’s ataxia, where gene silencing is an important feature leading to disease.Citation17,Citation18 However, there was no previous evidence that specific HDACs participate in the expansion process itself.
What is the evidence that HDACs are causal for expansions? The budding yeast HDACs Rpd3L and Hda1 were identified in a forward genetic screen for TNR promoting factors ().Citation15 This blind screen identified several subunits of Rpd3L and Hda1 at a frequency about 100-fold higher than random chance. Subsequent genetic analysis with targeted knockouts of Rpd3L or Hda1 showed suppression of expansion rates by up to 90%. In double mutants lacking both HDACs, the expansion phenotype was even more pronounced. Treatment of wild type yeast with the HDAC inhibitor TSA also suppressed expansions. This analysis in budding yeast indicated that both the class I enzyme Rpd3L and the class II prototype Hda1Citation19 promote TNR expansions.
Figure 1. Summary of HDACs and HATs described in the text.

Using a human astrocytic cell line, we showed that the class I enzyme HDAC3 also promotes expansions.Citation15 Knockdown with siRNA or treatment with an HDAC3-selective inhibitor blocked about 75% of expansions (). In contrast, knockdown or inhibition of HDAC1 did not suppress expansions. The involvement of HDACs in promoting expansions is consistent with an earlier report in Drosophila, where treatment of flies with TSA reduced expansion frequencies.Citation20 The Drosophila work also identified CBP as a histone acetyltransferase (HAT) with the contravening effect. CBP stabilized TNRs, based on the observation that CBP haploinsufficient animals displayed elevated expansion frequencies.Citation20 We knocked down human CBP, the related HAT p300, or both and saw increased frequencies of expansions in cultured astrocytes.Citation15 CBP and p300 are now implicated as factors that inhibit expansions in human cells (), thus stabilizing these repeating DNA elements.
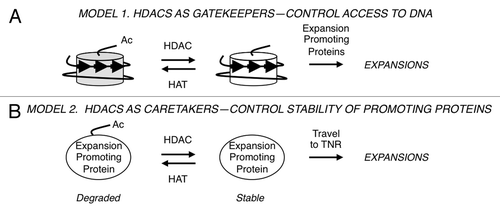
HDACs do not have the enzymatic capacity to alter DNA lengths, so it was clear that there must be downstream targets. Two types of models were tested in budding yeast for HDAC effects on triplet repeat expansions ().Citation15 A ‘gatekeeper’ hypothesis posits that Rpd3L and Hda1 modify histones near the TNR and thereby regulate access of the proteins that actually expand the DNA (). The gatekeeper would therefore exert its influence locally. To test this idea, the TNR reporter was integrated into two types of genomic location. In “hot” zones, transcript levelsCitation21,Citation22 and histone acetylationCitation23,Citation24 are exceedingly sensitive to the presence or absence of Rpd3. “Cold” zones, where message levels and histone acetylation are indifferent to Rpd3 status, were also tested. Surprisingly, there were no changes in either expansion rates or histone H4 pan-acetylation near the TNR regardless of whether the triplet repeat was integrated at hot or cold zones.Citation15 In budding yeast, the gatekeeper model is not supported by the available data. Instead, the results are better explained by a caretaker model (), where HDACs influence proteins or genes at a distance, which then travel to the TNR to exert their effects. Although transcriptional regulation is the most obvious caretaker mechanism, the data instead support HDAC stabilization of the non-histone protein Sae2, a nuclease. (Budding yeast Sae2 nuclease is unrelated to the human enzyme SAE2, which activates SUMO for post-translational SUMOylation of proteins.) In the caretaker model, yeast Sae2 represents an expansion promoting protein (). Sae2 is stabilized by deacetylation in an Rpd3- and Hda1-dependent manner, so in mutants lacking these HDACs, Sae2 is degraded.Citation25 As predicted by the caretaker model, mutants deficient in Sae2 or the related nuclease Mre11 also gave lower expansion rates, and double mutant analysis supported a pathway involving both Rpd3 and Mre11 ().Citation15 It is likely that Rpd3L and Hda1 target other proteins besides Sae2 that promote expansions in budding yeast, as loss of Sae2 gave a milder expansion phenotype than HDAC defects. Importantly, the mechanism in human cells—either gatekeeper or caretaker—awaits elucidation.
Figure 2. Speculative models of HDAC activity during triplet repeat expansions. (A) Gatekeeper hypothesis. The nucleosome structure at triplet repeats (arrows) and changes to histone tail acetylation are shown. Expansion promoting proteins are theorized to have improved access to the TNR DNA upon histone deacetylation. (B) Caretaker hypothesis. Stabilization of expansion promoting proteins (such as Sae2) by deacetylation allows these proteins more opportunity to travel to the trinucleotide repeat and catalyze the expansion.
How might Sae2 provoke expansions? Sae2 is most often thought of as an end-processing enzyme required for efficient repair of DNA double-strand breaks (detailed in the next section). However, expansions of short TNRs do not require other double-strand break repair proteins like Rad51 and Rad52.Citation26,Citation27 This distinction led to the suggestion that TNR hairpins, not double-strand breaks, are the relevant targets of Sae2 and Mre11 in our experimental system.Citation15 By this model, TNR hairpin processing requires Sae2 and Mre11 but cleavage does not lead to, or result from, a double-strand break. This idea is supported by the observations that Sae2 and Mre11 process DNA hairpins both in vivo and in vitro,Citation28,Citation29 and that virtually all models of TNR expansions envision a mutagenic intermediate comprised of a hairpin or other aberrant DNA structure.Citation1-Citation3 However, it remains to be proven that Sae2 and Mre11 actually process a TNR hairpin to promote expansions.
Double-Strand Break Repair
DNA double-strand breaks (DSBs) are one of the most serious types of DNA damage. Eukaryotic cells repair DSBs in two ways, one that is homology-dependent and a second process called non-homologous end joining (NHEJ). Both pathways rejoin the break and therefore stabilize the chromosome, although NHEJ often entails gains or losses of nucleotides at the break site and is therefore somewhat mutagenic. It is well known that HDACs and HATs are among the many factors recruited to DSBs.Citation30 In most cases, it was thought that HDACs act primarily as gatekeepers, helping to selectively recruit proteins to the DSB for repair and damage signaling.Citation30 Two recent studies provide a better understanding for how HDACs modulate DSB repair. In one case, specific HDACs act as caretakers,Citation25 whereas the second paper helps clarify a gatekeeper function by distinct HDACs.Citation31
The laboratory of Marco Foiani discovered that the budding yeast HDACs Rpd3 and Hda1 link processing of DSBs with the DNA damage response and autophagy.Citation25 Inhibition of Rpd3 and Hda1 or mutation of the genes encoding them resulted in inability to process the DSB for homology-dependent repair and also in poor activation of the DNA damage response. Thus, Rpd3 and Hda1 favor homology-dependent repair (). The relevant target of Rpd3 and Hda1 in this case was not histones, but rather the nucleases Sae2 and ExoI, which are necessary for processing of the broken chromosome ends. Yeast Sae2 and ExoI are deacetylated in an Rpd3- and Hda1-dependent manner, although it is not clear yet whether this deacetylation is direct. Deacetylation of the nucleases protects them from degradation via autophagy. The counteracting HAT is Gcn5, which acetylates Sae2 and ExoI (). The mammalian form of Sae2, called CtIP, is also deacetylated to promote end processing, although the relevant HDACs is SIRT6,Citation32 a class III enzyme. Nevertheless, it is now clear that Rpd3 and Hda1 are caretakers during homology-dependent repair of DSBs, helping avoid mutation by stabilizing key nucleases and allowing these enzymes to perform their important repair functions.Citation25 This is similar to the caretaker model shown in . In fact, the rationale for testing Sae2 in TNR expansions was inspired by the findings of Robert et al.Citation25
Non-homologous end joining is also regulated by specific HDACs, as highlighted in a recent publication from the laboratory of Steve Jackson.Citation31 NHEJ usually requires minimal processing of the DSB, occurs rapidly, and uses a discrete set of proteins, all of which set NHEJ apart from homology-dependent repair.Citation33 Miller et al. showed that HDAC1 and HDAC2 are the key HDACs in promoting NHEJ ().Citation31 HDAC1 and HDAC2 were rapidly recruited to sites of DNA damage induced by several agents that cause DSBs. Depletion of HDAC1 and HDAC2 caused cell hypersensitivity to DNA damaging agents and a sustained DNA damage response, consistent with reduced levels of NHEJ. HDAC1 and HDAC2 were important for globally deacetylating histones at H3K56 and H4K16, with the former modification also perturbed at sites of DNA damage. The authors note that depletion of HDAC1 and HDAC2 likely cause additional defects besides NHEJ,Citation31 so caution must be used to avoid oversimplifying the picture. Two gatekeeper-type models (similar to ) were put forth.Citation31 HDAC1 and HDAC2 may affect the ability of NHEJ factors to access DSBs or to function effectively there. Alternatively, HDAC1 and HDAC2 may act to repress transcription at sites of DNA damage, thus preventing transcription from interfering with repair. A partial contribution of a caretaker model, based on direct targeting of NHEJ factors by the HDACs, could not be ruled out, however.
Concluding Remarks
There are several surprises in the work summarized above. The first is that certain HDACs help cause mutagenesis at trinucleotide repeats. A second surprise is the specificity of the relevant HDACs. There are 18 known human HDACsCitation19,Citation34 but only a few were shown to be important for NHEJCitation31 or TNR expansions.Citation15 Third, HDACs serve as both caretakers and gatekeepers during DNA mutagenesis and repair by targeting both non-histone proteins and histones. Together, these findings underscore the importance of HDACs on genome stability.
Along with these new insights come unexpected challenges. In terms of mechanism, the multifaceted functions of HDACs make it difficult to tease out all the ways they regulate control DNA mutagenesis and repair. Gene expression, chromatin structure, and the firing of DNA replication origins are just some of the additional considerations that must be included in thinking about HDACs and the genome.Citation35-Citation39 With respect to medicine, the use of HDAC inhibitors (HDACi) as potential therapies has great promise,Citation40,Citation41 but only now are we beginning to understand the ramifications of these inhibitors and their possible side effects on genomic stability. For example, using HDACi to sensitize tumors to radiation could lead to secondary mutations.Citation31,Citation42 On the other hand, using HDACi to offset the transcriptional defects in Huntington’s diseaseCitation43 may have the additional benefit of reducing somatic expansions at the HD gene and thereby slowing disease progression.Citation15 For HDACi therapy to be optimally beneficial, it is crucial that specific HDACs continue to be identified in relevant disease-related pathways, and that isotype-selective inhibitors be further developed for more precise targeting.
Acknowledgments
Funded by Science Foundation Ireland grant 10/IN.1/B2973 to R.S.L., and by a postgraduate scholarship from the Irish Research Council for Science, Engineering and Technology to A.F. Space limitations precluded us from including all the exciting studies in this field, and we request those authors’ understanding.
Related Research Data
References
- Mirkin SM. Expandable DNA repeats and human disease. Nature 2007; 447:932 - 40; http://dx.doi.org/10.1038/nature05977; PMID: 17581576
- Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo.. Cell Res 2008; 18:198 - 213; http://dx.doi.org/10.1038/cr.2008.5; PMID: 18166978
- López Castel A, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 2010; 11:165 - 70; http://dx.doi.org/10.1038/nrm2854; PMID: 20177394
- Fu Y-H, Kuhl DPA, Pizzuti A, Pieretti M, Sutcliffe JS, Richards SR, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67:1047 - 58; http://dx.doi.org/10.1016/0092-8674(91)90283-5; PMID: 1760838
- Lahue RS, Slater DL. DNA repair and trinucleotide repeat instability. Front Biosci 2003; 8:s653 - 65; http://dx.doi.org/10.2741/1107; PMID: 12700078
- McMurray CT. Hijacking of the mismatch repair system to cause CAG expansion and cell death in neurodegenerative disease. DNA Repair (Amst) 2008; 7:1121 - 34; http://dx.doi.org/10.1016/j.dnarep.2008.03.013; PMID: 18472310
- Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet 1999; 23:471 - 3; http://dx.doi.org/10.1038/70598; PMID: 10581038
- Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet 2001; 27:407 - 11; http://dx.doi.org/10.1038/86906; PMID: 11279522
- van den Broek WJAA, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJTA, et al. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet 2002; 11:191 - 8; http://dx.doi.org/10.1093/hmg/11.2.191; PMID: 11809728
- Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, et al. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J 2003; 22:2264 - 73; http://dx.doi.org/10.1093/emboj/cdg202; PMID: 12727892
- Wheeler VC, Lebel L-A, Vrbanac V, Teed A, te Riele H, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet 2003; 12:273 - 81; http://dx.doi.org/10.1093/hmg/ddg056; PMID: 12554681
- Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet 2004; 13:1815 - 25; http://dx.doi.org/10.1093/hmg/ddh186; PMID: 15198993
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007; 447:447 - 52; http://dx.doi.org/10.1038/nature05778; PMID: 17450122
- Hubert L Jr., Lin Y, Dion V, Wilson JH. Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum Mol Genet 2011; 20:4822 - 30; http://dx.doi.org/10.1093/hmg/ddr421; PMID: 21926083
- Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol 2012; 10:e1001257; http://dx.doi.org/10.1371/journal.pbio.1001257; PMID: 22363205
- Dion V, Wilson JH. Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet 2009; 25:288 - 97; http://dx.doi.org/10.1016/j.tig.2009.04.007; PMID: 19540013
- Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol 2006; 2:551 - 8; http://dx.doi.org/10.1038/nchembio815; PMID: 16921367
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007; 30:575 - 621; http://dx.doi.org/10.1146/annurev.neuro.29.051605.113042; PMID: 17417937
- Yang X-J, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008; 9:206 - 18; http://dx.doi.org/10.1038/nrm2346; PMID: 18292778
- Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science 2007; 315:1857 - 9; http://dx.doi.org/10.1126/science.1139517; PMID: 17332375
- Bernstein BE, Tong JK, Schreiber SL. Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci U S A 2000; 97:13708 - 13; http://dx.doi.org/10.1073/pnas.250477697; PMID: 11095743
- Fazzio TG, Kooperberg C, Goldmark JP, Neal C, Basom R, Delrow J, et al. Widespread collaboration of Isw2 and Sin3-Rpd3 chromatin remodeling complexes in transcriptional repression. Mol Cell Biol 2001; 21:6450 - 60; http://dx.doi.org/10.1128/MCB.21.19.6450-6460.2001; PMID: 11533234
- Rundlett SE, Carmen AA, Suka N, Turner BM, Grunstein M. Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4 by RPD3. Nature 1998; 392:831 - 5; http://dx.doi.org/10.1038/33952; PMID: 9572144
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005; 123:581 - 92; http://dx.doi.org/10.1016/j.cell.2005.10.023; PMID: 16286007
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471:74 - 9; http://dx.doi.org/10.1038/nature09803; PMID: 21368826
- Miret JJ, Pessoa-Brandão L, Lahue RS. Orientation-dependent and sequence-specific expansions of CTG/CAG trinucleotide repeats in Saccharomyces cerevisiae.. Proc Natl Acad Sci U S A 1998; 95:12438 - 43; http://dx.doi.org/10.1073/pnas.95.21.12438; PMID: 9770504
- Bhattacharyya S, Lahue RS. Yeast Srs2 DNA helicase selectively blocks expansions of trinucleotide repeats. Mol Cell Biol 2004; 24:7324 - 30; http://dx.doi.org/10.1128/MCB.24.17.7324-7330.2004; PMID: 15314145
- Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 2002; 108:183 - 93; http://dx.doi.org/10.1016/S0092-8674(02)00614-1; PMID: 11832209
- Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell 2007; 28:638 - 51; http://dx.doi.org/10.1016/j.molcel.2007.11.001; PMID: 18042458
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409 - 33; http://dx.doi.org/10.1101/gad.2021311; PMID: 21363960
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 2010; 17:1144 - 51; http://dx.doi.org/10.1038/nsmb.1899; PMID: 20802485
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010; 329:1348 - 53; http://dx.doi.org/10.1126/science.1192049; PMID: 20829486
- Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J 2009; 417:639 - 50; http://dx.doi.org/10.1042/BJ20080413; PMID: 19133841
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 2010; 5:253 - 95; http://dx.doi.org/10.1146/annurev.pathol.4.110807.092250; PMID: 20078221
- Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. Histone acetylation regulates the time of replication origin firing. Mol Cell 2002; 10:1223 - 33; http://dx.doi.org/10.1016/S1097-2765(02)00702-5; PMID: 12453428
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693 - 705; http://dx.doi.org/10.1016/j.cell.2007.02.005; PMID: 17320507
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 2007; 76:75 - 100; http://dx.doi.org/10.1146/annurev.biochem.76.052705.162114; PMID: 17362198
- Knott SRV, Viggiani CJ, Tavaré S, Aparicio OM. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev 2009; 23:1077 - 90; http://dx.doi.org/10.1101/gad.1784309; PMID: 19417103
- Unnikrishnan A, Gafken PR, Tsukiyama T. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 2010; 17:430 - 7; http://dx.doi.org/10.1038/nsmb.1780; PMID: 20228802
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26:5420 - 32; http://dx.doi.org/10.1038/sj.onc.1210610; PMID: 17694083
- Gray SG. Epigenetic treatment of neurological disease. Epigenomics 2011; 3:431 - 50; http://dx.doi.org/10.2217/epi.11.67; PMID: 22126204
- Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett 2009; 274:169 - 76; http://dx.doi.org/10.1016/j.canlet.2008.06.005; PMID: 18635312
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A 2008; 105:15564 - 9; http://dx.doi.org/10.1073/pnas.0804249105; PMID: 18829438