Abstract
Fibrosis of any tissue is characterized by excessive extracellular matrix accumulation that ultimately destroys tissue architecture and eventually abolishes normal organ function. Although much research has focused on the mechanisms underlying disease pathogenesis, there are still no effective antifibrotic therapies that can reverse, stop or delay the formation of scar tissue in most fibrotic organs. As fibrosis can be described as an aberrant wound healing response, a recent hypothesis suggests that the cells involved in this process gain an altered heritable phenotype that promotes excessive fibrotic tissue accumulation. This article will review the most recent observations in a newly emerging field that links epigenetic modifications to the pathogenesis of fibrosis. Specifically, the roles of DNA methylation and histone modifications in fibrotic disease will be discussed.
Introduction
Fibrotic disease comprises a wide array of clinical entities that are thought to account for approximately 45% of all deaths in the developed world.Citation1 Although fibrosis can affect numerous organs, and is likely triggered by a variety of injurious insults, the core pathways underlying scar tissue formation are likely to be similar in any organ type.Citation2 Despite this, there are currently no effective anti-fibrotic therapies available to treat fibrotic disease. Recently, epigenetic mechanisms have been proposed as playing a role in promoting a heritable pro-fibrotic phenotype in cells involved in scar tissue formation. Understanding the epigenetic control of cellular phenotype in the context of tissue injury and aberrant tissue remodeling could in the future lead to the development of epigenetic therapies to target fibrosis.
Fibrosis and the Role of Fibroblasts
Fibrosis is characterized by excessive extracellular matrix deposition that destroys normal tissue architecture and eventually disrupts organ function. Development of fibrosis is attributed to a variety of causes including persistent infections, autoimmune reactions, allergic responses, chemical insults, radiation, and tissue injury. Although there are a variety of insults that can cause fibrosis, it is the fibroblast that generates the collagen-rich scar tissue. In normal healthy tissue, resident fibroblasts maintain the structural integrity of an organ by synthesizing the scaffolding that supports normal tissue architecture. At rest fibroblasts synthesize relatively low amounts of extracellular matrix but they must be able to respond to injurious insults in order to produce the provisional matrix that is involved in normal wound repair. It can thus be argued that fibroblasts must have a very high degree of phenotypic plasticity for efficient wound repair and normal tissue remodeling. Upon activation, fibroblasts differentiate into myofibroblasts (). If unregulated, as is the case during fibrosis, this differentiated cell type can be responsible for the pathological accumulation of extracellular matrix proteins and may ultimately lead to loss of organ function. Myofibroblasts can be distinguished from fibroblasts as they express high levels of cytokines, express the contractile cytoskeleton protein α-smooth muscle actin (αSMA) and synthesize increased levels of extracellular matrix proteins including collagens.Citation3,Citation4 During fibrosis the numbers of myofibroblasts within the diseased tissue increases. Evidence suggests that these cells can be derived from several sources including, (1) increased proliferation of resident fibroblasts, (2) tissue infiltration of circulating fibrocytes, (3) epithelial/endothelial mesenchymal transition (EMT/EndMT), whereby resident epithelial/endothelial cells lose their cell markers and gain a mesenchymal phenotype enabling them to effectively act like a fibroblast and contribute to the fibrotic milieu in a similar manner as resident fibroblasts.Citation1 However, it should be noted that the precise contribution, if any, of EMT/EndMT to the fibroblast pool in fibrotic disease is a topic of contentious debate at the present.Citation5
Figure 1. Fibroblast to myofibroblast differentiation and the role of epigenetic modifications Following injury, fibroblasts differentiate into myofibroblasts. This process is encouraged by pro-fibrotic stimulants, such as transforming growth factor β (TGFβ), inflammatory cytokines and hypoxia. Under normal circumstances, wound-healing myofibroblasts contribute to tissue remodeling and repair and are eventually removed by apoptosis. In contrast, fibrotic myofibroblasts are hyperproliferative and continually secrete pro-fibrotic cytokines and collagens into their surrounding environment. These cells are resistant to apoptosis and are therefore continually present in the tissue. As this review highlights, DNA hypermethylation as well as histone modifications have been implicated in controlling this process. Hence, the use of DNA methyltransferase inhibitors (DNMTi) or histone deacetylase inhibitors (HDACi) may be beneficial in preventing myofibroblast differentiation.
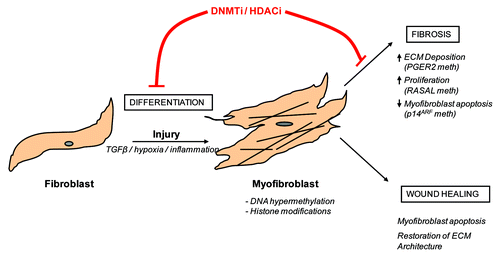
Regardless of the source of activated fibroblasts, tissue fibrosis can be described as a pathological process during which there is a persistent change in fibroblast phenotype to a myofibroblast that promotes disease progression. Fibroblast to myofibroblast differentiation occurs during normal wound healing. However, when the injured tissue is repaired, myofibroblasts disappear through apoptosis.Citation6 This is in contrast to “fibrotic” myofibroblasts which resist apoptosis. Thus, it is apparent that a permanent, stable change occurs in “fibrotic” myofibroblasts, which promotes their persistence in fibrotic tissue. A switch to a hyperactive, apoptosis-resistant myofibroblast is supported by in vitro evidence, which has demonstrated that myofibroblasts extracted from fibrotic tissue retain their hyperactive phenotype, despite the fact that they are no longer growing in a pro-fibrotic environment.Citation7-Citation10 Thus, a role for epigenetic regulation of myofibroblast differentiation has been proposed as a potential mechanism in regulating this process. Given the interest and availability of epigenetic therapies in the treatment of cancer, fully understanding and investigating these mechanisms in fibrosis may lead to the discovery of future epigenetic-based anti-fibrotic therapies.
Epigenetics and Disease
Epigenetic modifications play an essential role in processes such as development and differentiation; however, there is also strong evidence linking epigenetics to the pathogenesis of a variety of diseases.Citation11 A role for epigenetics in cancer development is well acknowledgedCitation12 and epigenetic therapies are now considered as treatment options for certain cancers. The demethylating agent 5-azacytidine (5-aza; Vidaza) and its analog 5-aza-2'-deoxycytidine (5-azadC; Decitabine) are already approved for treatment of myelodysplastic syndromes (MDS).Citation13 Histone modifying drugs have also been approved for the treatment of cancer, for example, the histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA) is used for treatment of cutaneous T cell lymphoma.Citation14
Although cancer is the most studied disease with relevance to epigenetic modifications, it is likely that alterations in the epigenome may play important roles in the development of other disease pathologies. Recently, roles for both DNA methylation and histone modifications in fibrosis have been reported.
Global DNA Methylation and Fibrosis
Global DNA methylation analysis studies have demonstrated that significant alterations in DNA methylation occur in fibrotic tissues. Rabinovich et al. compared levels of 5-methyl-cytosine (5-mC) in idiopathic pulmonary fibrosis (IPF), lung cancer and normal lung tissue samples and recorded significant changes in the CpG island methylation profile between the groups. IPF samples displayed global hypomethylation compared with normal tissue samples, which resembled the profile of adenocarcinoma samples.Citation15 In support of a role for DNA methylation-related modifications in IPF, Sanders et al. quantified the expression of the DNA methyltransferases (DNMTs) in IPF tissue and demonstrated increased mRNA expression of de novo enzymes in disease as well as increased DNMT3a protein expression.Citation16 Global analysis of DNA methylation in a murine model of liver fibrosis implicated hypomethylation in diseased tissue when compared with normal liver specimens.Citation17 Although studies quantifying CpG methylation levels in heterogeneous tissue implicated alterations in DNA methylation as a participant in fibrosis pathogenesis, cell-type specific global 5-mC alterations remain largely unknown. It is likely that the methylation profile of specific cell types is masked when global whole tissue analysis is performed. In fact, numerous fibroblast studies have implicated a hypermethylated profile in these cells as promoting myofibroblast differentiation and, thus, disease progression.
DNA Methylation and Myofibroblast Differentiation
Various studies have alluded to gene-specific hypermethylation events as contributing to myofibroblast differentiation. Huang et al. used IPF patient-derived fibroblasts and fibroblasts from a bleomycin mouse model of pulmonary fibrosis, both of which express low/no prostaglandin E receptor 2 (PTGER2). PTGER2 loss promoted fibrosis and the group demonstrated that this was due to promoter hypermethylation.Citation18 The group also investigated the global levels of DNA methylation in normal and fibrotic mouse and human pulmonary fibroblasts and observed that genomic DNA of the fibrotic cells was hypermethylated compared with normal control fibroblasts.Citation18Fibroblasts are a heterogeneous cell population both in vivo and in vitro. In vitro fibroblasts can be separated into two distinct populations based on expression of a glycoprotein called Thy-1. Thy-1 negative fibroblasts are associated with a more fully differentiated myofibroblast phenotype when compared with Thy-1 positive cells.Citation19Hagood et al. reported that in areas of active fibrosis (fibroblastic foci) in IPF patient lung tissue there was loss of Thy-1.Citation20 Loss of Thy-1 was shown to be due to Thy-1 promoter methylation.Citation20 DNA methylation has also been implicated in the pathogenesis of renal fibrosis. Bechtel et al. used a folic acid-induced renal fibrosis model in mice to explore the role of DNA methylation and the therapeutic efficacy of the non-specific DNMT inhibitor, 5-aza. They demonstrated that the extent of fibrosis in animals treated with 5-aza was significantly reduced when compared with control animals.Citation21 Modifications in DNA methylation were mediated by an increase in DNMT1 expression in the folic acid challenged mice that promoted hypermethylation. DNMT1+/− mice had significantly reduced kidney fibrosis in this model.Citation21 The group also suggested that the fibrotic response induced by folic acid treatment was due, at least in part, to hypermethylation of the Ras GTPase activating-like protein 1 (RASAL1) promoter in kidney fibroblasts and that this helped promote a hyperactive myofibroblast phenotype.Citation21 Other studies have also correlated RASAL1 methylation and expression to myofibroblast differentiation. Li et al. focused on liver fibrosis. This group treated rat hepatic stellate cells, which normally undergo myofibroblast transdifferentiation that includes loss of RASAL1, with 5-azadC, which blocked the decrease in its expression.Citation22 Further evidence of a role for gene-specific methylation and fibrosis is provided by Wang and colleagues. The authors reported that hypermethylation of the collagen I gene repressor Fli-1 occurs in scleroderma fibroblasts and implied that this may impact on the over production of collagen within the multi-organ fibrotic disease.Citation23
p14ARF is a tumor suppressor protein and when upregulated it induces cell cycle arrest.Citation21 Given that IPF fibroblasts are considered to be resistant to apoptosis, it is interesting to note that one group has recently demonstrated an increased incidence of hypermethylation of the p14ARF promoter in IPF fibroblasts.22This was associated with a apoptotic resistant cell phenotype.
Increased DNA methylation causes gene silencing via either one of two mechanisms. First, increased levels of 5-mC can physically prevent transcription factor binding thus repressing gene transcription. Second, and perhaps more importantly, methylated DNA recruits methyl binding domain proteins (MBDs), which results in chromatin remodeling via the recruitment of histone modifying enzymes. A specific MBD protein that is now linked to fibrosis is methyl CpG binding protein 2 (MeCP2). In vitro cultured hepatic stellate cells undergo differentiation to hepatic fibroblasts. Expression of MeCP2 increases accordingly and participates in promoting gene silencing, such as suppressing peroxisome proliferator-activated receptor gamma (PPARγ) and RASAL1, which oppose myofibroblast differentiation.Citation22,Citation24,Citation25 In fibrotic liver, expression of MeCP2 is selectively detected at actively fibrotic regions of tissue where myofibroblasts reside.Citation24 Additionally in a MeCP2 knockout mouse model of CCl4 liver fibrosis there was attenuation of numerous fibrotic factors.Citation25 The differentiation from hepatic stellate cell to myofibroblast can be prevented in vitro by treatment with 5-azadC, implying that a change in DNA methylation with subsequent MeCP2 binding is necessary for the differentiation.Citation24 A role for MeCP2 in pulmonary fibrosis has also been proposed. Suppression of MeCP2 expression in pulmonary fibroblasts increases αSMA expression which is a classical feature of myofibroblast differentiation.Citation26 Furthermore, in a bleomycin-induced pulmonary fibrosis model MeCP2−/− displayed decreased pulmonary collagen deposition and decreased myofibroblast differentiation compared with wild type animals.Citation26 While MeCP2 can bind to unmethylated DNA, it is well recognized that it preferentially binds to methylated DNA to assist and promote chromatin remodeling.Citation27 Thus, several independent studies linking MeCP2 to myofibroblast differentiation all implicate alterations in DNA methylation at the heart of this process.
In summary, the current literature strongly suggests that a hypermethylated fibroblast phenotype encourages myofibroblast differentiation; the numerous gene-specific methylation events that likely drive this change in phenotype, however, are yet to be fully described. The DNMTs likely play a crucial role in regulating this process; however, the individual roles of each of the DNMTs have yet to be properly defined in fibrotic disease.
Histone Modifications
The N-terminal tails of histone proteins can become modified through various mechanisms. These post-translational modifications, such as acetylation or methylation, can change the overall structure of the surrounding chromatin and thus have an impact on gene expression. Acetylation of lysine residues on histone tails can occur via enzymes called histone acetyltransferases (HATs). Acetylation of histone proteins can also be reversed by histone deacetylases (HDACs). Addition of methyl groups can cause amino acids of histones to become mono-, di-or trimethylated, a process that is mediated by histone methyltransferases (HMTs). As with acetylation, histone methylation is reversible. This process is governed by histone demethylases of which there are two main families: lysine specific histone demethyalase-1 (LSD1) and the jumonji domain (JmjC) containing histone demethylases. Histone modifications have also been implicated in the development and pathogenesis of fibrotic disease.
Histone Modifications and Fibrosis
A collection of publications have highlighted the significance of histone modifications within fibrotic diseases. For example, one study showed that in IPF fibroblasts, decreased cytokine-induced cyclooxygenase-2 (COX2) expression was due to defective histone acetylation, with decreased recruitment of HATs and increased recruitment of HDAC containing complexes to the COX2 promoter.Citation28 In a separate study, also in IPF fibroblasts, the authors found that dysregulation of the same histone modification pathways also suppressed expression of the CXC chemokine gamma interferon (IFN-γ)-inducible protein of 10 kDa (IP-10).Citation29 In a unilateral urethral obstruction (UUO) model in mice that results in renal fibrosis, the expression of HDAC1 and HDAC2 were increased relative to expression in the contralateral healthy kidney.Citation30 Hsp70-Hsp90 Organizing Protein (HOP) is a co-chaperone protein that can modulate cardiac growth by forming a complex with HDAC that inhibits transcriptional activity of serum response factor in cardiomyocytes. In mice, overexpression of HOP resulted in cardiac hypertrophy and interstitial fibrosis, which was rescued by histone deacetylase inhibitors (HDACi).Citation31 These studies implicate aberrant histone acetylation in pulmonary, renal and cardiac fibrosis. These support the hypothesis that such epigenetic mechanisms play an important role in the pathogenesis of fibrosis in any organ.
In further support of a role for histone acetylation in fibrotic disease, various HDACi have significant anti-fibrotic effects, where they have been shown to be effective at reversing myofibroblast differentiation. In human lung fibroblasts treated with the pro-fibrotic stimulant transforming growth factor β1 (TGFβ1), the HDACi SAHA reversed all pro-fibrotic effects of TGFβ1 assessed.Citation32 In a similar study treatment with trichostatin A (TSA), abrogated TGFβ1-induced myofibroblast differentiation,Citation33 while the class I specific HDACi, spiruchostatin A (SpA), also inhibited pulmonary myofibroblast differentiation.Citation34 Treating Thy-1 negative pulmonary fibroblasts with TSA caused re-expression of the protein further highlighting a role for histone modifications in regulating fibroblast phenotype.Citation35 In fibroblasts extracted from patients with systemic sclerosis, a disease characterized by fibrosis of the skin as well as various internal organs, treatment with TSA reduced cytokine-induced mRNA and protein expression of collagen I.Citation36 TSA has also been used to prevent myofibroblast differentiation in rat liver stellate cells that normally contribute to liver fibrosis. Additionally, Chen et al. have shown anti-fibrotic effects of TSA using alternative tissue culture models of fibroblasts that are proposed to mimic the natural crowded state of cells in tissue. Here, TSA suppressed TGFβ1-induced expression of collagen I as well as reducing cell proliferation.Citation37
The anti-fibrotic effects of HDACi have also been reported in in vivo models of fibrosis. Brown et al. treated a DOCA salt-induced rat model of systemic hypertension that is subsequently accompanied by cardiac fibrosis with SAHA. One of the main findings of this study was that animals treated with SAHA had decreased cardiac remodeling. The drug attenuated numerous fibrotic processes including increased inflammatory cytokine expression, collagen deposition and left ventricular hypertrophy.Citation38 In a mouse model of dermal fibrosis, which utilizes topically applied bleomycin, TSA abolished the bleomycin-induced increase in dermal thickness, demonstrating an interaction between TSA and extracellular matrix synthesis.Citation36 TSA also abrogated fibrosis in a UUO model of renal fibrosis.Citation39
Limited studies have investigated the contributions of histone methylation and fibrosis. During hepatic stellate cell differentiation there is increased expression of the histone methyltransferases Absent Small or Homeotic disk 1 (ASH1) that promotes expression of TGFβ, the Tissue Inhibitor of Matrix Metalloproteinase (TIMP) 1 and collagen I.Citation40 Similarly, in an ischemic renal injury model that causes fibrosis, changes in histone methyltransferases activity were recorded.Citation41
Epigenetic-Based Therapies in Fibrosis
There is strong evidence to suggest a role for both alterations in DNA methylation as well as histone modifications in fibrotic disease. Therefore, it is increasingly apparent that drugs targeting such epigenetic alterations will be beneficial in the treatment of fibrosis. As has been previously described, these agents have significant anti-fibrotic effects both in vivo and in vitro. It is also particularly encouraging that these agents are already approved for the treatment of certain cancers and are relatively well tolerated in humans.
It should be noted, however, that there are some issues with the use of these drugs as anti-fibrotic agents. Although studies suggest that HDACi may be an effective anti-fibrotic therapy, it should be noted that there have been recent reports linking TSA to emphysema and aberrant cardiac remodeling. HDACi have been associated with decreasing the expression of hypoxia inducible factors.Citation42 One group has demonstrated that treatment of adult rats with TSA caused partial emphysema by inhibiting hypoxia inducible factor 1-α (HIF1α), vascular endothelial growth factor (VEGF) and lysyl oxidase (LOX) expression.Citation43 While HDACi have been shown to have advantageous effects by preventing left ventricle remodeling and dysfunction in a chronic model of left ventricular pressure overload,Citation38 their effects on adaptation to acute right ventricle adaptation to pressure overload, as occurs in during pulmonary hypertension, is not beneficial. Treatment of pulmonary artery banded rats with broad spectrum HDACi resulted in right ventricle remodeling that was worse than in untreated animals.Citation44 The authors provided evidence that this may be due to the anti-angiogenic and/or pro-apoptotic effects of broad spectrum HDACi.Citation44 Hence, the use of HDACi could lead to severe unwanted side effects in situations where an adaptive angiogenic response to acute injury is required.
The majority of current studies have used HDACi and DNMT inhibitors that are non-specific and target all cell types. Given that the precise contribution of DNMTs and HDACs in fibrotic disease has not yet been fully elucidated, sustained treatment with these agents may lead to unwanted side effects. This may in the future warrant the design of more specific inhibitors, for example DNMT3a or 3b inhibitors could have beneficial effects by sparing DNMT1 activity and allowing a degree of maintenance DNA methylation in replicating cells of normal tissues.
While considerable effort is being directed toward the design of more specific HDAC and DNMT inhibitors, it is perhaps also of benefit to investigate the injury-related drivers of epigenetic modifications as these may also serve as effective drug targets. Thus, understanding the mechanisms that regulate epigenetic alterations is vital if we are to combat chronic fibrotic disease.
Factors that are Involved in Fibrosis that Could Regulate Epigenetic Modifications
Although there is strong evidence that epigenetic modifications play a role in the pathogenesis of fibrosis, the factors that regulate these epigenetic alterations have yet to be properly investigated and defined. Research efforts to decipher these mechanisms may be enhanced through application of what is already known in the more intensively studied field of cancer biology. There are some aspects of these two pathologies that are similar. One common feature between fibrosis and cancer is the potent cytokine TGFβ and its enhanced cell signaling through the SMAD family of proteins. In certain cancers, altered expression and activity of TGFβ is linked with tumorigenesis, including aberrant cell proliferation and apoptotic responses. Whereas in fibrotic pathologies increased TGFβ signaling is one of the key drivers of pathological accumulation of collagen, resulting in impaired organ function. In cancer research, TGFβ has been associated with alterations in the epigenetic profile of cells. For example, in prostate cancer cells and in an in vivo model, TGFβ1 was shown to regulate expression of DNMT1, the maintenance DNMT as well as the de novo DNMTs, 3a and 3b. Blockade of TGFβ signaling significantly decreased expression of all three DNMTs via a p-ERK mediated pathway.Citation45 TGFβ treatment has also been shown to alter the methylation status of the tumor suppressor gene p15. This active demethylation event involves loss of a repressor complex consisting of numerous proteins, including HDAC1/2 and DNMT3a and the co-recruitment of SMAD2/3, CBP and a DNA glycosylase.Citation46 Further evidence of TGFβ regulation of epigenetic changes is provided by a study linking prolonged TGFβ signaling with increased DNA methylation correlating with suppression of miR-200. This is also accompanied with sustained zinc finger E-box-binding homeobox (ZEB) expression.Citation47 The authors highlight the importance of this for switching between epithelial and mesenchymal phenotypic states, a phenomenon that may also be important in fibrosis. A role for DNA methylation in governing the TGFβ response in fibrosis is also provided by work highlighting TGFβ1-induced myofibroblast differentiation is greatly enhanced or reduced by knockdown or overexpression of DNA methyltransferases.Citation48 In addition, a recent publication has implicated TGFβ2 in the reduction of ADAM33 gene expression in lung fibroblasts through altering chromatin structure and DNA methylation.Citation49
In relation to histone modifications, TGFβ mediated suppression of interleukin -2 (IL-2) in immune cells was shown to occur by H3K9 trimethylation at the promoter region of the gene.Citation50 TGFβ1 treatment has also been shown to lead to histone methylation alterations at extracellular matrix associated gene promoters.Citation51
Other candidates that have been suggested in the cancer field to promote changes in epigenetic marks that may subsequently render the cell capable of encouraging disease progression include interleukin-6 (IL-6) or environmental factors such as hypoxia. Similar pathophysiological mechanisms may be relevant in fibrogenesis and are discussed below.
IL-6 has been implicated in many fibrotic processes. One example of this was highlighted by a study that infused IL-6 for one week into rats resulting in cardiac fibrosis and hypertrophy.Citation52 Of great relevance to the fibrotic field is the reported ability of IL-6 to regulate DNMT expression. Studies using cancer models have shown that IL-6 treatment can upregulate DNMT1, and this can be mediated by miR-148a and miR-152.Citation53,Citation54 Additional mechanisms by which IL-6 can drive DNMT1 expression include the transcription factor Fli-1.Citation55
There is also strong evidence to support a role for hypoxia in fibrosis pathogenesis.Citation56-Citation59 Hypoxia occurs when a cells demand for oxygen exceeds supply. Of note, hypoxia has been shown to regulate DNA methylation as well as modulate histone modifications.Citation60 A role for hypoxia in modulating the DNA methylation profile of cells has been described and is suggested to be due to hypoxic regulation of DNMT expression.Citation61-Citation63 Interestingly, there are putative hypoxia responsive elements (HRE) on the promoters of all three active DNMTs although the functional relevance of these remains to be investigated. Similarly there is good evidence that histone modifications can also be influenced by hypoxia. Hypoxic regulation of histone methyltransferases and deacetylases have both been reportedCitation61,Citation64-Citation68 In fact, one study has recently suggested that hypoxia-induced EMT is reliant upon histone modifications.Citation69 Further research is required to determine whether such hypoxia related epigenetic changes occur in fibrotic disease.
Conclusion
There is growing in vitro and in vivo evidence in the literature supporting the proposal that epigenetic therapeutics may be effective in the treatment of fibrosis. It is striking that such similar epigenetic control pathways appear to be active in all tissues studied and that targeting them may be an effective way of reducing myofibroblast differentiation and collagen deposition regardless of organ tested. However, there is limited knowledge about the exact epigenetic alterations that occur in fibrotic tissue either on a global or cell-specific scale that promote disease progression. Any drugs that have displayed anti-fibrotic properties have wide spanning epigenetic targets. Hence, little is known about gene-specific alterations that occur in fibrotic tissue and this certainly warrants further investigation. The underlying mechanisms that may promote changes in the epigenetic profile of these cells also needs to be fully elucidated. Investigating epigenetic pathways and fully understanding these mechanisms may lead to the use of epigenetic therapeutics as anti-fibrotic agents.
| Abbreviations | ||
| /Acronyms | = | αSMA, α-smooth muscle actin |
| EMT | = | epithelial mesenchymal transition |
| 5-azadC | = | 5-aza-2'-deoxycytidine (decitabine) |
| HDACi | = | histone deacetylase inhibitor |
| SAHA | = | suberoylanilide hydroxamic acid |
| 5-mC | = | 5-methyl-cytosine |
| DNMTs | = | DNA methyltransferases |
| MBDs | = | methyl binding domain proteins |
| TGFβ | = | transforming growth factor β |
| TSA | = | Trichostatin A |
| HRE | = | hypoxia response element |
Acknowledgments
The authors would like to thank Science Foundation Ireland and Health Research Board for funding.
Related Research Data
References
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199 - 210; http://dx.doi.org/10.1002/path.2277; PMID: 18161745
- Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: expressway to the core of fibrosis. Nat Med 2011; 17:552 - 3; http://dx.doi.org/10.1038/nm0511-552; PMID: 21546973
- Phan SH. Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc 2008; 5:334 - 7; http://dx.doi.org/10.1513/pats.200708-146DR; PMID: 18403329
- Darby IA, Hewitson TD. Fibroblast differentiation in wound healing and fibrosis. Int Rev Cytol 2007; 257:143 - 79; http://dx.doi.org/10.1016/S0074-7696(07)57004-X; PMID: 17280897
- Grgic I, Duffield JS, Humphreys BD. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol 2012; 27:183 - 93; http://dx.doi.org/10.1007/s00467-011-1772-6; PMID: 21311912
- Desmoulière A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995; 146:56 - 66; PMID: 7856739
- Jordana M, Schulman J, McSharry C, Irving LB, Newhouse MT, Jordana G, et al. Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis 1988; 137:579 - 84; PMID: 3345039
- Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 2005; 183:225 - 37; http://dx.doi.org/10.1007/s00408-004-2534-z; PMID: 16211459
- Rodemann HP, Müller GA. Abnormal growth and clonal proliferation of fibroblasts derived from kidneys with interstitial fibrosis. Proc Soc Exp Biol Med 1990; 195:57 - 63; PMID: 2399261
- Rodemann HP, Müller GA. Characterization of human renal fibroblasts in health and disease: II. In vitro growth, differentiation, and collagen synthesis of fibroblasts from kidneys with interstitial fibrosis. Am J Kidney Dis 1991; 17:684 - 6; PMID: 2042650
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429:457 - 63; http://dx.doi.org/10.1038/nature02625; PMID: 15164071
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27 - 36; http://dx.doi.org/10.1093/carcin/bgp220; PMID: 19752007
- Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006; 106:1794 - 803; http://dx.doi.org/10.1002/cncr.21792; PMID: 16532500
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007; 12:1247 - 52; http://dx.doi.org/10.1634/theoncologist.12-10-1247; PMID: 17962618
- Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, et al. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci U S A 2000; 97:1778 - 83; http://dx.doi.org/10.1073/pnas.97.4.1778; PMID: 10677534
- Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA Methylation Profile in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2012; In press http://dx.doi.org/10.1164/rccm.201201-0077OC; PMID: 22700861
- Komatsu Y, Waku T, Iwasaki N, Ono W, Yamaguchi C, Yanagisawa J. Global analysis of DNA methylation in early-stage liver fibrosis. BMC Med Genomics 2012; 5:5; http://dx.doi.org/10.1186/1755-8794-5-5; PMID: 22281153
- Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC, et al. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol 2010; 177:2245 - 55; http://dx.doi.org/10.2353/ajpath.2010.100446; PMID: 20889571
- Sanders YY, Kumbla P, Hagood JS. Enhanced myofibroblastic differentiation and survival in Thy-1(-) lung fibroblasts. Am J Respir Cell Mol Biol 2007; 36:226 - 35; http://dx.doi.org/10.1165/rcmb.2006-0178OC; PMID: 16960126
- Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol 2008; 39:610 - 8; http://dx.doi.org/10.1165/rcmb.2007-0322OC; PMID: 18556592
- Bechtel W, McGoohan S, Zeisberg EM, Müller GA, Kalbacher H, Salant DJ, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med 2010; 16:544 - 50; http://dx.doi.org/10.1038/nm.2135; PMID: 20418885
- Tao H, Huang C, Yang JJ, Ma TT, Bian EB, Zhang L, et al. MeCP2 controls the expression of RASAL1 in the hepatic fibrosis in rats. Toxicology 2011; 290:327 - 33; http://dx.doi.org/10.1016/j.tox.2011.10.011; PMID: 22056649
- Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum 2006; 54:2271 - 9; http://dx.doi.org/10.1002/art.21948; PMID: 16802366
- Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ 2007; 14:275 - 85; http://dx.doi.org/10.1038/sj.cdd.4401979; PMID: 16763620
- Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, Tsukamoto H, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010; 138:705 - 14, 714, e1-4; http://dx.doi.org/10.1053/j.gastro.2009.10.002; PMID: 19843474
- Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. Am J Pathol 2011; 178:1500 - 8; http://dx.doi.org/10.1016/j.ajpath.2011.01.002; PMID: 21435439
- Nan X, Tate P, Li E, Bird A. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol 1996; 16:414 - 21; PMID: 8524323
- Coward WR, Watts K, Feghali-Bostwick CA, Knox A, Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol 2009; 29:4325 - 39; http://dx.doi.org/10.1128/MCB.01776-08; PMID: 19487460
- Coward WR, Watts K, Feghali-Bostwick CA, Jenkins G, Pang L. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol Cell Biol 2010; 30:2874 - 86; http://dx.doi.org/10.1128/MCB.01527-09; PMID: 20404089
- Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol 2010; 298:F133 - 41; http://dx.doi.org/10.1152/ajprenal.00400.2009; PMID: 19906951
- Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest 2003; 112:863 - 71; PMID: 12975471
- Wang Z, Chen C, Finger SN, Kwajah S, Jung M, Schwarz H, et al. Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis?. Eur Respir J 2009; 34:145 - 55; http://dx.doi.org/10.1183/09031936.00084808; PMID: 19224893
- Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol 2009; 297:L864 - 70; http://dx.doi.org/10.1152/ajplung.00128.2009; PMID: 19700647
- Davies ER, Haitchi HM, Thatcher TH, Sime PJ, Kottmann RM, Ganesan A, et al. Spiruchostatin A inhibits proliferation and differentiation of fibroblasts from patients with pulmonary fibrosis. Am J Respir Cell Mol Biol 2012; 46:687 - 94; http://dx.doi.org/10.1165/rcmb.2011-0040OC; PMID: 22246864
- Sanders YY, Tollefsbol TO, Varisco BM, Hagood JS. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts. Am J Respir Cell Mol Biol 2011; 45:16 - 23; http://dx.doi.org/10.1165/rcmb.2010-0154OC; PMID: 20724553
- Diao JS, Xia WS, Yi CG, Wang YM, Li B, Xia W, et al. Trichostatin A inhibits collagen synthesis and induces apoptosis in keloid fibroblasts. Arch Dermatol Res 2011; 303:573 - 80; http://dx.doi.org/10.1007/s00403-011-1140-1; PMID: 21400246
- Chen CZ, Peng YX, Wang ZB, Fish PV, Kaar JL, Koepsel RR, et al. The Scar-in-a-Jar: studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br J Pharmacol 2009; 158:1196 - 209; http://dx.doi.org/10.1111/j.1476-5381.2009.00387.x; PMID: 19785660
- Iyer A, Fenning A, Lim J, Le GT, Reid RC, Halili MA, et al. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br J Pharmacol 2010; 159:1408 - 17; http://dx.doi.org/10.1111/j.1476-5381.2010.00637.x; PMID: 20180942
- Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, et al. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 2009; 297:F996 - 1005; http://dx.doi.org/10.1152/ajprenal.00282.2009; PMID: 19640900
- Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, et al. The histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012; In press; http://dx.doi.org/10.1002/hep.25754; PMID: 22488473
- Zager RA, Johnson AC. Renal ischemia-reperfusion injury upregulates histone-modifying enzyme systems and alters histone expression at proinflammatory/profibrotic genes. Am J Physiol Renal Physiol 2009; 296:F1032 - 41; http://dx.doi.org/10.1152/ajprenal.00061.2009; PMID: 19261745
- Chen S, Sang N. Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotechnol 2011; 2011:197946; http://dx.doi.org/10.1155/2011/197946; PMID: 21151670
- Mizuno S, Yasuo M, Bogaard HJ, Kraskauskas D, Natarajan R, Voelkel NF. Inhibition of histone deacetylase causes emphysema. Am J Physiol Lung Cell Mol Physiol 2011; 300:L402 - 13; http://dx.doi.org/10.1152/ajplung.00207.2010; PMID: 21224215
- Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, et al. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 2011; 183:1402 - 10; http://dx.doi.org/10.1164/rccm.201007-1106OC; PMID: 21297075
- Zhang Q, Chen L, Helfand BT, Jang TL, Sharma V, Kozlowski J, et al. TGF-β regulates DNA methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and predicts disease recurrence. PLoS One 2011; 6:e25168; http://dx.doi.org/10.1371/journal.pone.0025168; PMID: 21980391
- Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, Tini M, et al. TGF-β-Dependent Active Demethylation and Expression of the p15(ink4b) Tumor Suppressor Are Impaired by the ZNF217/CoREST Complex. Mol Cell 2012; 46:636 - 49; http://dx.doi.org/10.1016/j.molcel.2012.03.027; PMID: 22560925
- Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell 2011; 22:1686 - 98; http://dx.doi.org/10.1091/mbc.E11-02-0103; PMID: 21411626
- Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Epigenetic regulation of myofibroblast differentiation by DNA methylation. Am J Pathol 2010; 177:21 - 8; http://dx.doi.org/10.2353/ajpath.2010.090999; PMID: 20489138
- Yang Y, Wicks J, Haitchi HM, Powell RM, Manuyakorn W, Howarth PH, et al. Regulation of a disintegrin and metalloprotease-33 expression by transforming growth factor-β. Am J Respir Cell Mol Biol 2012; 46:633 - 40; http://dx.doi.org/10.1165/rcmb.2011-0030OC; PMID: 22227561
- Wakabayashi Y, Tamiya T, Takada I, Fukaya T, Sugiyama Y, Inoue N, et al. Histone 3 lysine 9 (H3K9) methyltransferase recruitment to the interleukin-2 (IL-2) promoter is a mechanism of suppression of IL-2 transcription by the transforming growth factor-β-Smad pathway. J Biol Chem 2011; 286:35456 - 65; http://dx.doi.org/10.1074/jbc.M111.236794; PMID: 21862595
- Sun G, Reddy MA, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol 2010; 21:2069 - 80; http://dx.doi.org/10.1681/ASN.2010060633; PMID: 20930066
- Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010; 56:225 - 31; http://dx.doi.org/10.1161/HYPERTENSIONAHA.109.148635; PMID: 20606113
- Foran E, Garrity-Park MM, Mureau C, Newell J, Smyrk TC, Limburg PJ, et al. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res 2010; 8:471 - 81; http://dx.doi.org/10.1158/1541-7786.MCR-09-0496; PMID: 20354000
- Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010; 51:881 - 90; PMID: 20146264
- Hodge DR, Xiao W, Clausen PA, Heidecker G, Szyf M, Farrar WL. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem 2001; 276:39508 - 11; http://dx.doi.org/10.1074/jbc.C100343200; PMID: 11551897
- Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int 2000; 58:2351 - 66; http://dx.doi.org/10.1046/j.1523-1755.2000.00419.x; PMID: 11115069
- Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, et al. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med 2007; 176:1108 - 19; http://dx.doi.org/10.1164/rccm.200705-683OC; PMID: 17761615
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 2007; 117:3810 - 20; PMID: 18037992
- Short M, Nemenoff RA, Zawada WM, Stenmark KR, Das M. Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am J Physiol Cell Physiol 2004; 286:C416 - 25; http://dx.doi.org/10.1152/ajpcell.00169.2003; PMID: 14561589
- Watson JA, Watson CJ, McCann A, Baugh J. Epigenetics, the epicenter of the hypoxic response. Epigenetics 2010; 5:293 - 6; http://dx.doi.org/10.4161/epi.5.4.11684; PMID: 20418669
- Watson JA, Watson CJ, McCrohan AM, Woodfine K, Tosetto M, McDaid J, et al. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Hum Mol Genet 2009; 18:3594 - 604; http://dx.doi.org/10.1093/hmg/ddp307; PMID: 19584087
- Shahrzad S, Bertrand K, Minhas K, Coomber BL. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007; 2:119 - 25; http://dx.doi.org/10.4161/epi.2.2.4613; PMID: 17965619
- Skowronski K, Dubey S, Rodenhiser D, Coomber B. Ischemia dysregulates DNA methyltransferases and p16INK4a methylation in human colorectal cancer cells. Epigenetics 2010; 5:547 - 56; http://dx.doi.org/10.4161/epi.5.6.12400; PMID: 20543577
- Tausendschön M, Dehne N, Brüne B. Hypoxia causes epigenetic gene regulation in macrophages by attenuating Jumonji histone demethylase activity. Cytokine 2011; 53:256 - 62; http://dx.doi.org/10.1016/j.cyto.2010.11.002; PMID: 21131212
- Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, et al. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun 2008; 372:892 - 7; http://dx.doi.org/10.1016/j.bbrc.2008.05.150; PMID: 18538129
- Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 2010; 30:344 - 53; http://dx.doi.org/10.1128/MCB.00444-09; PMID: 19858293
- Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 2008; 283:36542 - 52; http://dx.doi.org/10.1074/jbc.M804578200; PMID: 18984585
- Yang J, Ledaki I, Turley H, Gatter KC, Montero JC, Li JL, et al. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann N Y Acad Sci 2009; 1177:185 - 97; http://dx.doi.org/10.1111/j.1749-6632.2009.05027.x; PMID: 19845621
- Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell 2011; 43:811 - 22; http://dx.doi.org/10.1016/j.molcel.2011.07.012; PMID: 21884981