Abstract
Mature microRNAs (miRNAs) are a class of small non-coding RNAs involved in posttranslational gene silencing. Previous studies found that downregulation of miRNAs is a common feature observed in solid tumors, including hepatocellular carcinoma (HCC). We employed a genome-wide approach to test the hypothesis that DNA methylation alterations in miRNA host genes may cause deregulated miRNA expression in HCC. We analyzed tumor and adjacent non-tumor tissues from 62 Taiwanese HCC cases using Infinium HumanMethylation27 DNA Analysis BeadChips that include 254 CpG sites covering 110 miRNAs from 64 host genes. Expression levels of three identified miRNAs (miR-10a, miR-10b and miR-196b) were measured in a subset of 37 HCC tumor and non-tumor tissues. After Bonferroni adjustment, a total of 54 CpG sites from 27 host genes significantly differed in DNA methylation levels between tumor and adjacent non-tumor tissues with 53 sites significantly hypermethylated in tumor tissues. Among the 54 significant CpG sites, 15 sites had more than 2-fold tumor/non-tumor changes, 17 sites had differences > 10%, and 10 sites had both features [including 8 significantly hypermethylated CpG sites in the host genes of miR-10a, miR-10b and miR-196b (HOXB4, HOXD4 and HOXA9, respectively)]. Significant downregulation of miR-10a was observed in tumor compared with non-tumor tissues (0.50 vs. 1.73, p = 0.031). The concordance for HOXB4 methylation alteration and dysregulation of miR-10a was 73.5%. No significant change was observed for miR-10b expression. Unexpectedly, miR-196b was significantly upregulated in tumor compared with non-tumor tissues (p = 0.0001). These data suggest that aberrant DNA methylation may lead to dysregulation of miR-10a in HCC tumor tissues.
Keywords: :
Introduction
Hepatocellular carcinoma (HCC) is a common and aggressive cancer with an estimated 748,300 new cases and 695,900 deaths worldwide in 2008.Citation1 Hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are the major etiological risk factors, accounting for 80% of all HCC cases. Environmental exposures to alcohol, aflatoxins and cigarette smoke have also been implicated.Citation2 It is known that the initiation or development of HCC is a multi-step process involving accumulated genetic and epigenetic alterations resulting from carcinogenic exposures. Such alterations can lead to the onset of tumorigenesis by mediating the dysregulation of relevant genes. However, while aberrant DNA methylation in protein-coding genes, a type of epigenetic modification, has been frequently found in HCC tumor tissues,Citation3 correlations with corresponding changes in expression of the relevant genes were not always consistent,Citation4-Citation7 indicating the necessity of measuring gene expression to clarify the biological functions of aberrant DNA methylation. However, it is not technically feasible using biospecimens currently available for most clinical or population studies.
Regulation of gene expression can also be mediated by microRNAs (miRNAs), a class of small (18–26 nucleotide) non-protein-coding RNAs that are stable and can be measured in paraffin samples. miRNAs function as negative regulators of the expression of over 60% of protein-coding genes by accelerating the degradation of target mRNA and by translational inhibition.Citation8 A variety of tumorigenic processes, including cellular proliferation, differentiation and apoptosis,Citation9-Citation13 involve the participation of miRNAs. Previous studies revealed that the expression of certain miRNAs was significantly downregulated in HCC tumor compared with non-tumor, cirrhotic and normal liver tissues,Citation14-Citation21 suggesting the potential tumor suppressive role of miRNA host genes. miRNAs are usually nested within their host genes and aberrant epigenetic alterations occurring in these regions may cause dysregulation of the relevant mature miRNAs. Previous studies explored miRNA methylation and malignancy mainly using animal models and cancer cell lines.Citation22-Citation26 A few miRNAs (miR-1, miR-191, miR-124, miR-125b and miR-203) are silenced in HCC tissues through a DNA methylation mechanism.Citation27-Citation30 However, the available data measuring both DNA methylation and miRNA expression alterations in human HCC tissues are very limited.
In the current study, we employed a genome-wide approach to test the hypothesis that DNA methylation alterations in miRNA host genes may cause deregulated mature miRNA expression in HCC. If significantly different methylation patterns are observed between HCC tumor and adjacent non-tumor tissues that are mostly cirrhotic, they are more likely to be related to malignant transformation than to be the result of cirrhosis. If the DNA methylation patterns significantly correlate with repression of the relevant miRNAs, the biological effects should be greater than the silencing of a single protein-coding gene, since a single miRNA can regulate multiple downstream target genes. These amplified effects indicate that miRNAs may be potentially more sensitive biomarkers to distinguish early malignant transformation and improve HCC diagnosis and prognosis. We previously reported on the analysis of 62 paired tumor and adjacent non-tumor tissues from Taiwanese HCC cases using the Infinium HumanMethylation27 DNA Analysis BeadChip identifying the CpG sites that most significantly differed by tissue status.Citation31 Here, we evaluate the 254 CpG sites on the array that cover 110 miRNAs from 64 host genes. We first identified aberrant miRNA gene methylation in HCC tumor tissue, and then examined the expression changes of several identified miRNAs.
Results
miRNA gene methylation differentiates HCC tumor from non-tumor tissues
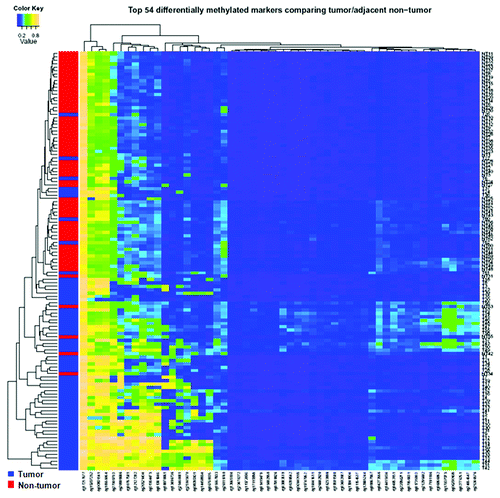
We compared the methylation levels between 62 tumor and adjacent non-tumor paired tissues at each of the 254 miRNA related autosomal CpG sites. After Bonferroni adjustment, 54 out of 254 sites in 27 genes significantly differed in methylation levels, with 53 sites significantly hypermethylated in tumor tissues (Table S1), which may lead to downregulation of the corresponding mature miRNAs. But most miRNAs loci (78.7%) showed no significant difference of DNA methylation between tumor and non-tumor tissues. Among the 54 significant CpG sites, 15 sites have > 2-fold tumor/non-tumor methylation changes; 30 sites have > 1.5-fold tumor/non-tumor methylation changes; 17 sites have a > 10% difference in methylation levels between tumor and non-tumor tissues; and 10 sites have both > 2-fold tumor/non-tumor changes and > 10% difference (), including eight significantly hypermethylated CpG sites in HOXB4, HOXD4 and HOXA9—the host genes of miR-10a, miR-10b and miR-196b, respectively. displays mean DNA methylation differences between the 62 paired tumor and adjacent non-tumor tissues at all 254 CpG sites using a volcano plot. A total of 54 CpG sites were significantly different with –log10 p values ≤ 0.05 after Bonferroni correction. Hierarchical clustering of the 54 significant CpG sites was performed to differentiate tumor and non-tumor tissues (). In general, good separation of tumor and adjacent tissues was obtained with a small amount of misclassification (5 tumors and 8 non-tumor tissues misclassified). Among the top ten significant miRNA host genes with over 1.5-fold tumor/non-tumor methylation changes, three genes (HOXB4, HOXD4 and HOXA9) with available TaqMan miRNA expression assays (miR-10a, miR-10b and miR-196b) were selected for determination of the concordance between DNA methylation and miRNA expression. DNA methylation differences between paired tumor and non-tumor tissue for the three host genes are shown in Figure S1. The hypermethylation rates in HCC tumor tissues were 90.3%, 74.2% and 77.4% for HOXB4, HOXD4 and HOXA9, respectively.
Table 1. The 22 significant CpG sites covering 10 miRNA host genes with tumor and adjacent non-tumor methylation differences > 0.10 or fold changes > 2.0 ranked by statistical significance
Figure 1. Volcano plot for differential DNA methylation analysis of 254 miRNA CpG sites in 62 paired HCC tumor and adjacent non-tumor tissues. The x-axis shows the mean DNA methylation difference, while the y-axis shows the –log10 of the adjusted p value for each CpG site, representing the strength of association. Above the solid line indicates statistically significant (p ≤ 0.05) after Bonferroni correction.
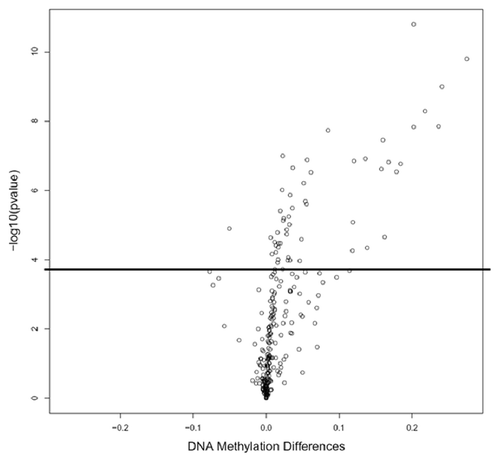
Figure 2. Hierarchical cluster analysis of all 54 significantly differentially methylated miRNA CpG sites between 62 tumor and adjacent non-tumor tissues. Blue represents tumor tissue, and red represents adjacent non-tumor tissue.
Enriched biological pathways of significant miRNA host genes
PANTHER ontology analysis conducted for all 27 significant miRNA host genes to identified four enriched biological pathways, including Wnt signaling, Ras, glutamine/glutamate conversion, oxidative stress response, and mannose metabolism pathways. Two pathways (glutamine/glutamate conversion and mannose metabolism) were statistically significant after Bonferroni correction for multiple comparisons (p ≤ 0.05) (Table S2).
Aberrant DNA methylation in miRNA host gene is correlated with HCC risk factors
The methylation differences between tumor and non-tumor tissues were compared for miRNA genes by HCC risk factors, including viral status, AFB1-DNA adduct levels, alcohol drinking and cigarette smoking (Table S3). Overall, only a few CpG sites of miRNA genes differentially methylated in tumor and adjacent non-tumor tissues are potentially related to HCC risk factors after Bonferroni correction. The difference between DNA methylation of tumor and adjacent non-tumor tissues significantly differed for PIGO (HS_104) by viral status (positive vs. negative) and for ZNF207 (miR-632) by alcohol drinking status (never vs. ever). Differences in methylation for other miRNA host genes did not differ by HCC risk factors.
miRNAs expression in HCC tumor/non-tumor tissues and correlations with DNA methylation levels
Expression levels of three miRNAs (miR-10a, miR-10b and miR-196b) were measured based on the availability of appropriate assays. Their genomic locations and the distances to relevant host genes are shown in Figure S2. The three miRNAs are located on chromosome 17q21.32, 2q31.1 and 7p15.2, respectively. All are at the upstream of their host genes. The distances to their host genes (HOXB4, HOXD4 and HOXA9) are 1.46 kb, 0.97 kb and 3.95 kb, respectively. Expression of the three miRNAs was measured in a subset of 37 HCC tumor and non-tumor tissues (). Significant downregulation of miR-10a expression (mean: 0.50 vs. 1.73, p = 0.031) was found in tumor compared with non-tumor tissues, consistent with the pattern of host gene HOXB4 hypermethylation in tumors. Although not statistically significant, higher expression of miR-10b was observed in tumor (3.59) compared with non-tumor tissues (0.21, p = 0.308, ). While miR-196b expression was significantly upregulated in HCC tumor compare with non-tumor tissues, the overall expression levels in both tumor and non-tumor tissues were very low (0.03 vs. 0.004, p = 0.0001).
Table 2. miRNA expression comparisons between HCC tumor and adjacent non-tumor tissues (37 pairs)
Among 29 paired tumor/non-tumor tissues with both DNA methylation and miRNA expression data, the concordances between DNA methylation alterations (HOXB4, HOXD4 and HOXA9) and miRNAs’ dysregulation (miR-10a, miR-10b and miR-196b) are shown in Figure S3. A good concordance of 75.9% was observed for HOXB4 methylation alteration and miR-10a dysregulation. Pearson correlation analysis showed consistently negative correlations between miR-10a expression and methylation at all 5 CpG sites of miR-10a’s host gene (r ranges from -0.102 to -0.197), but none achieved statistical significance (Table S4). No significant correlations were found between miR-10b expression and methylation levels of the two relevant CpG sites (data not shown). The correlation between miR-196b expression and the averaged methylation level over the four relevant CpG sites was 0.525 (p < 0.0001), opposite to the expected negative correlation. The concordance between HOXD4 and HOXA9 methylation alterations and dysregulation of miR-10b and miR-196b were very low from 34.5% to 17.2% (Fig. S3).
Discussion
We identified a total of 54 CpG sites in 27 miRNAs host genes, including three HOX family members that were significantly hypermethylated in HCC tumor compared with non-tumor tissues (). We further confirmed that miR-10a expression was significantly downregulated in HCC tumor tissues (), consistent with the hypermethylation of its host gene (HOXB4). A good concordance of 75.9% was also observed for HOXB4 methylation alteration and miR-10a dysregulation (Fig. S3). These preliminary data suggest that aberrant CpG methylation in the promoter region of HOXB4 might be responsible for silencing of miR-10a expression in HCC tumorigenesis.
miR-10a is located upstream of its HOXB4 host gene on chromosome 17q21.32, where several CpG islands are situated within a 3-kb distance. A previous study found aberrant DNA methylation of HOXB4 in extrahepatic cholangiocarcinoma.Citation32 Downregulation miR-10a expression has been found in cancer cell lines and solid tumor tissues, including in prostate,Citation33 colon,Citation34 esophageal squamous cell carcinoma,Citation35 and head and neck squamous cell carcinoma.Citation36 Similar under-expression of miR-10a was also observed in malignantly transformed human bronchial epithelial cells.Citation37 Repressed miR-10a could be restored by treatment with 5-aza-2'-deoxycytidine (AZA), a demethylating agent,Citation37 or by double knockout of DNMT1 and DNMT3b,Citation34 indicating its epigenetic regulation. These results strongly support our observation that hypermethylated CpG sites in HOXB4 might cause under-expression of miR-10a and failure to inhibit down-stream target oncogenes.
Other supporting evidence comes from previous studies on miR-10a downstream target genes. miR-10a has six experimentally verified target genes (βTRC, HOXA1, HOXD4, MAP3K7, NCOR2 and USF2)Citation38,Citation39 and two putative targets (HOXA3 and HOXD10).Citation34 Four targets belong to the homeobox family containing transcription factors that play a pivotal role during normal growth and differentiation of mammalian tissues. In addition to being significantly upregulated in a malignantly transformed human bronchial epithelial cell line,Citation37 HOXA1 mRNA was also upregulated in non-small cell lung cancer.Citation40 Forced expression of HOXA1 resulted in dramatically increased anchorage-independent proliferation in immortalized human mammary epithelial cells.Citation41 HOXA1 mediated oncogenic transformation is through promotion of cell survival by transcriptional upregulation of Bcl-2, abrogation of the apoptotic response,Citation41 or modulation of the P44/42 MAP kinase pathway.Citation42 Expression of another homeobox family member, HOXD4, was also suppressed by miR-10a in breast, cervical, HCC and lung adenocarcinoma cell lines.Citation38 miR-10a, as a posttranscriptional regulator of the IκB/NF-κB signaling pathway, can suppress βTRC and MAP3K7.Citation43 Knockdown of miR-10a led to activation of NF-κB signaling transduction and elevation of pro-inflammation.Citation44 These data suggest that miR-10a acts as a tumor suppressor and, when repressed, there may be decreased targeting of downstream oncogenes that results in increased cell proliferation, migration, invasiveness or angiogenesis,Citation35 decreased apoptosis or dedifferentiation,Citation40 ultimately leading to tumor formation.Citation45
However, several studies have also identified an oncogenic role for miR-10a, which was significantly overexpressed in pancreatic cancer,Citation46 neuroblastomaCitation47 and, in contrast to our results, in HCC.Citation48,Citation49 The reasons for these differences are not clear. Vrba et al. suggested an epigenetic control mechanism for miRNAs since they observed a small set of miRNAs (including miR-10a and miR-10b) with dual epigenetic repressive marks where both DNA methylation and H3K27me3 were present at their promoters. Disruption of one epigenetic mechanism may be insufficient to initiate aberrant expression of miRNAs.Citation50 Another explanation may be different data normalizers used in different studies (U6 snRNA in the current study vs. global median or miR-140).Citation48,Citation49 Therefore, further studies are warranted to elucidate other potential mechanisms that lead to different miR-10a expression patterns in HCC.
The strengths of the current study include the use of genome-wide methylation array screening of miRNA host genes in the largest set of HCC cases studied to date and the evaluation of the effects of methylation alterations by measuring expression of several relevant miRNAs. Among the limitations is the small sample size in the subgroup with both DNA methylation and miRNA expression data. Only a few aberrant methylation markers were identified as associated with viral status or alcohol drinking status due to the high frequency of viral infection and alcohol drinking limiting power. In addition, the heterogeneity of tissue materials (fresh or FFPE) used for measurements of methylation and miRNA, respectively, may have impacted our analysis of potential relationships between DNA methylation alterations and dysregulated miRNAs expression. However, several previous studies have compared DNA methylation or miRNAs expression patterns between FFPE and fresh tissues.Citation51-Citation54 Overall, a good correlation was obtained (correlation coefficients ranged from 0.81 to 0.95), indicating the potential influence of different sample types on epigenetic biomarkers was minimal. Because the Infinium HumanMethylation27 array did not cover CpG sites in genomic regions of many dysregulated miRNAs identified in previous HCC studies (such as miR-199a, miR-21, miR-301 and miR-122),Citation49,Citation55 we could not directly examine correlations between DNA methylation and relevant miRNAs expression for these miRNAs. An expanded study using Infinium HumanMethylation450K arrays that cover 3,440 miRNAs is ongoing and may help to more comprehensively understand the role of aberrant DNA methylation in miRNA dysregulation in a genome-wide scale.
In conclusion, we found that DNA methylation alterations frequently occur in miRNA host genes in HCC. Decreased miR-10a expression in tumor tissues was related to its host gene’s hypermethylation, supporting a tumor suppressor role for miR-10a in the repression of downstream target oncogenes. Therefore, aberrant miRNA methylation and expression markers may be useful molecular biomarkers for assessing HCC risk and early diagnosis. Further studies are warranted to explore other biological mechanisms involved in miRNAs regulation in HCC.
Methods
Patients and biopsy specimens
This study was approved by Institutional Review Boards of Columbia University and National Taiwan University (NTU). Written informed consent was obtained.
Patients’ demographic data and clinicopathologic characteristics have been reported previously.Citation31 Briefly, 62 frozen liver tissues collected in the Department of Surgery, NTU Hospital were kept at −70°C until shipment to Columbia University where pathologic analysis confirmed HCC status and indicated that adjacent tissues were primarily cirrhotic. Since fresh tissue was depleted, a subset of 37 paired formalin-fixed, paraffin-embedded (FFPE) HCC tumor and non-tumor tissue sections were used to detect miRNA expression.
DNA preparation and Illumina Infinium Human Methylation platform
The details of the approach has been previously described.Citation31 The complete set of Infinium HumanMethylation27 data has been deposited in NCBI’s Gene Expression Omnibus (GEO)Citation56 and are available through series accession number GSE37988 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37988).
RNA extraction and miRNA quantification
Total RNA was extracted from one 5 µm FFPE tissue section using RecoverAll Total Nucleic Acid Isolation Kit (Applied Biosystems, Foster City, CA) as per the manufacturer's protocol. TaqMan® MicroRNA Assays (Applied Biosystems) were used to quantitate miRNA expression levels. A simple, two-step protocol requires only 10 ng miRNA per reverse transcription reaction with a miRNA-specific primer (TaqMan MicroRNA Reverse Transcription Kit, Applied Biosystems), followed by real-time PCR with TaqMan probes. PCR was performed on an Applied Biosystems 7500 Sequence Detection System. PCR reaction mixtures contained 1.33 ul RT reaction product and Universal PCR Master Mix in a total volume of 20 ul. Cycling variables are as follows: 95°C for 10 min followed by 40 cycles at 95°C (15 sec) and annealing/extension at 60°C (1 min). All reactions were performed in duplicate. The expression levels of target miRNAs were normalized to the endogenous controls (U6 snRNA), and calculated as 2-∆Ct values. The difference of miRNA expression between tumor and non-tumor tissue was displayed as log2-transformed fold-change.
Statistical analysis
β values were generated using the Illumina BeadStudio software.Citation57 Sites on the sex chromosomes were removed from the analysis leaving 254 CpG sites relevant to miRNA genes. Paired t-tests with Bonferroni correction for multiple testing were used to identify CpG sites that were differentially methylated between tumor and adjacent non-tumor tissues. A significant difference was defined as sites with a Bonferroni-corrected p value ≤ 0.05. A volcano plot displayed mean DNA methylation differences for all 254 CpG sites. Hierarchical clustering of the methylation data was performed with the significant CpG sites. Gene ontology analysis was performed by the PANTHER classification system (www.pantherdb.org) to compare the significantly methylated gene list with the reference (NCBI, human genome build 36).Citation58 The binomial test was used to identify significantly enriched pathways, biological processes, molecular functions, cellular components and protein class terms after Bonferroni correction for multiple comparisons with a cutoff of p ≤ 0.05. Two-sample t-test was used to investigate if methylation differences in miRNAs between tumor and adjacent non-tumor tissues are affected by HCC risk factors, such as viral (HBV, HCV) status (positive vs. negative), aflatoxin B1 (AFB1) DNA adduct level (high vs. low),Citation59 alcohol consumption (ever vs. never), and cigarette smoking (ever vs. never). P values with and without Bonferroni correction are shown. These analyses were conducted using R language (www.r-project.org/). Paired t-test was used to examine difference of miRNA expression between tumor and adjacent non-tumor tissue. Pearson correlation coefficients were calculated to evaluate the relationship between miRNA expression and DNA methylation. The statistical analyses were completed using Statistical Analysis System 9.0 (SAS Institute).
| Abbreviations: | ||
| AFB1 | = | aflatoxin B1 |
| miRNA | = | microRNA |
| FFPE | = | formalin-fixed, paraffin-embedded |
| GEO | = | Gene Expression Omnibus |
| HBV | = | 1hepatitis B virus |
| HCV | = | hepatitis C virus |
| HCC | = | hepatocellular carcinoma |
| TSS | = | transcription start sites |
Additional material
Download Zip (371.9 KB)Acknowledgments
This work was supported by NIH grants R01 ES005116, P30 ES009089, R03 CA156629, R03 CA150140 and P30 CA013696. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/22140
Related Research Data
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69 - 90; http://dx.doi.org/10.3322/caac.20107; PMID: 21296855
- Chuang SC, La Vecchia C, Boffetta P. Liver cancer: descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett 2009; 286:9 - 14; http://dx.doi.org/10.1016/j.canlet.2008.10.040; PMID: 19091458
- Mínguez B, Lachenmayer A. Diagnostic and prognostic molecular markers in hepatocellular carcinoma. Dis Markers 2011; 31:181 - 90; PMID: 22045404
- Yasen M, Obulhasim G, Kajino K, Mogushi K, Mizushima H, Tanaka S, et al. DNA binding protein A expression and methylation status in hepatocellular carcinoma and the adjacent tissue. Int J Oncol 2012; 40:789 - 97; PMID: 22159460
- Pereira B, Oliveira C, David L, Almeida R. CDX2 promoter methylation is not associated with mRNA expression. Int J Cancer 2009; 125:1739 - 42; http://dx.doi.org/10.1002/ijc.24544; PMID: 19569237
- Shah N, Lin B, Sibenaller Z, Ryken T, Lee H, Yoon JG, et al. Comprehensive analysis of MGMT promoter methylation: correlation with MGMT expression and clinical response in GBM. PLoS One 2011; 6:e16146; http://dx.doi.org/10.1371/journal.pone.0016146; PMID: 21249131
- Formeister EJ, Tsuchiya M, Fujii H, Shpyleva S, Pogribny IP, Rusyn I. Comparative analysis of promoter methylation and gene expression endpoints between tumorous and non-tumorous tissues from HCV-positive patients with hepatocellular carcinoma. Mutat Res 2010; 692:26 - 33; http://dx.doi.org/10.1016/j.mrfmmm.2010.07.013; PMID: 20736025
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15 - 20; http://dx.doi.org/10.1016/j.cell.2004.12.035; PMID: 15652477
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281 - 97; http://dx.doi.org/10.1016/S0092-8674(04)00045-5; PMID: 14744438
- Pasquinelli AE, Hunter S, Bracht J. MicroRNAs: a developing story. Curr Opin Genet Dev 2005; 15:200 - 5; http://dx.doi.org/10.1016/j.gde.2005.01.002; PMID: 15797203
- Carleton M, Cleary MA, Linsley PS. MicroRNAs and cell cycle regulation. Cell Cycle 2007; 6:2127 - 32; http://dx.doi.org/10.4161/cc.6.17.4641; PMID: 17786041
- Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med 2009; 60:167 - 79; http://dx.doi.org/10.1146/annurev.med.59.053006.104707; PMID: 19630570
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6:857 - 66; http://dx.doi.org/10.1038/nrc1997; PMID: 17060945
- Braconi C, Patel T. MicroRNA expression profiling: a molecular tool for defining the phenotype of hepatocellular tumors. Hepatology 2008; 47:1807 - 9; http://dx.doi.org/10.1002/hep.22326; PMID: 18506877
- Huang YS, Dai Y, Yu XF, Bao SY, Yin YB, Tang M, et al. Microarray analysis of microRNA expression in hepatocellular carcinoma and non-tumorous tissues without viral hepatitis. J Gastroenterol Hepatol 2008; 23:87 - 94; http://dx.doi.org/10.1111/j.1440-1746.2007.05223.x; PMID: 18171346
- Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology 2008; 47:1955 - 63; http://dx.doi.org/10.1002/hep.22256; PMID: 18433021
- Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res 2007; 67:6092 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-4607; PMID: 17616664
- Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W, et al. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 2006; 99:671 - 8; http://dx.doi.org/10.1002/jcb.20982; PMID: 16924677
- Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, et al. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006; 25:2537 - 45; http://dx.doi.org/10.1038/sj.onc.1209283; PMID: 16331254
- Wong QW, Lung RW, Law PT, Lai PB, Chan KY, To KF, et al. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology 2008; 135:257 - 69; http://dx.doi.org/10.1053/j.gastro.2008.04.003; PMID: 18555017
- Li W, Xie L, He X, Li J, Tu K, Wei L, et al. Diagnostic and prognostic implications of microRNAs in human hepatocellular carcinoma. Int J Cancer 2008; 123:1616 - 22; http://dx.doi.org/10.1002/ijc.23693; PMID: 18649363
- Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 2006; 5:2220 - 2; http://dx.doi.org/10.4161/cc.5.19.3340; PMID: 17012846
- Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 2007; 6:1001 - 5; http://dx.doi.org/10.4161/cc.6.9.4209; PMID: 17457051
- Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9:435 - 43; http://dx.doi.org/10.1016/j.ccr.2006.04.020; PMID: 16766263
- Saito Y, Friedman JM, Chihara Y, Egger G, Chuang JC, Liang G. Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem Biophys Res Commun 2009; 379:726 - 31; http://dx.doi.org/10.1016/j.bbrc.2008.12.098; PMID: 19116145
- Kunej T, Godnic I, Ferdin J, Horvat S, Dovc P, Calin GA. Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat Res 2011; 717:77 - 84; http://dx.doi.org/10.1016/j.mrfmmm.2011.03.008; PMID: 21420983
- Alpini G, Glaser SS, Zhang JP, Francis H, Han Y, Gong J, et al. Regulation of placenta growth factor by microRNA-125b in hepatocellular cancer. J Hepatol 2011; 55:1339 - 45; http://dx.doi.org/10.1016/j.jhep.2011.04.015; PMID: 21703189
- Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res 2008; 68:5049 - 58; http://dx.doi.org/10.1158/0008-5472.CAN-07-6655; PMID: 18593903
- Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010; 31:766 - 76; http://dx.doi.org/10.1093/carcin/bgp250; PMID: 19843643
- Huang J, Wang Y, Guo Y, Sun S. Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology 2010; 52:60 - 70; http://dx.doi.org/10.1002/hep.23660; PMID: 20578129
- Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology 2012; 55:1799 - 808; http://dx.doi.org/10.1002/hep.25569; PMID: 22234943
- Shu Y, Wang B, Wang J, Wang JM, Zou SQ. Identification of methylation profile of HOX genes in extrahepatic cholangiocarcinoma. World J Gastroenterol 2011; 17:3407 - 19; http://dx.doi.org/10.3748/wjg.v17.i29.3407; PMID: 21876633
- Waltering KK, Porkka KP, Jalava SE, Urbanucci A, Kohonen PJ, Latonen LM, et al. Androgen regulation of micro-RNAs in prostate cancer. Prostate 2011; 71:604 - 14; http://dx.doi.org/10.1002/pros.21276; PMID: 20945501
- Han L, Witmer PD, Casey E, Valle D, Sukumar S. DNA methylation regulates MicroRNA expression. Cancer Biol Ther 2007; 6:1284 - 8; PMID: 17660710
- Matsushima K, Isomoto H, Kohno S, Nakao K. MicroRNAs and esophageal squamous cell carcinoma. Digestion 2010; 82:138 - 44; http://dx.doi.org/10.1159/000310918; PMID: 20588024
- Hui AB, Lenarduzzi M, Krushel T, Waldron L, Pintilie M, Shi W, et al. Comprehensive MicroRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res 2010; 16:1129 - 39; http://dx.doi.org/10.1158/1078-0432.CCR-09-2166; PMID: 20145181
- Shen YL, Jiang YG, Greenlee AR, Zhou LL, Liu LH. MicroRNA expression profiles and miR-10a target in anti-benzo[a] pyrene-7, 8-diol-9, 10-epoxide-transformed human 16HBE cells. Biomed Environ Sci 2009; 22:14 - 21; http://dx.doi.org/10.1016/S0895-3988(09)60016-7; PMID: 19462682
- Tan Y, Zhang B, Wu T, Skogerbø G, Zhu X, Guo X, et al. Transcriptional inhibiton of Hoxd4 expression by miRNA-10a in human breast cancer cells. BMC Mol Biol 2009; 10:12; http://dx.doi.org/10.1186/1471-2199-10-12; PMID: 19232136
- Agirre X, Jiménez-Velasco A, San José-Enériz E, Garate L, Bandrés E, Cordeu L, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res 2008; 6:1830 - 40; http://dx.doi.org/10.1158/1541-7786.MCR-08-0167; PMID: 19074828
- Abe M, Hamada J, Takahashi O, Takahashi Y, Tada M, Miyamoto M, et al. Disordered expression of HOX genes in human non-small cell lung cancer. Oncol Rep 2006; 15:797 - 802; PMID: 16525661
- Zhang X, Zhu T, Chen Y, Mertani HC, Lee KO, Lobie PE. Human growth hormone-regulated HOXA1 is a human mammary epithelial oncogene. J Biol Chem 2003; 278:7580 - 90; http://dx.doi.org/10.1074/jbc.M212050200; PMID: 12482855
- Mohankumar KM, Xu XQ, Zhu T, Kannan N, Miller LD, Liu ET, et al. HOXA1-stimulated oncogenicity is mediated by selective upregulation of components of the p44/42 MAP kinase pathway in human mammary carcinoma cells. Oncogene 2007; 26:3998 - 4008; http://dx.doi.org/10.1038/sj.onc.1210180; PMID: 17213808
- Fang Y, Shi C, Manduchi E, Civelek M, Davies PF. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci U S A 2010; 107:13450 - 5; http://dx.doi.org/10.1073/pnas.1002120107; PMID: 20624982
- Qin B, Yang H, Xiao B. Role of microRNAs in endothelial inflammation and senescence. Mol Biol Rep 2012; 39:4509 - 18; http://dx.doi.org/10.1007/s11033-011-1241-0; PMID: 21952822
- Hernando E. microRNAs and cancer: role in tumorigenesis, patient classification and therapy. Clin Transl Oncol 2007; 9:155 - 60; http://dx.doi.org/10.1007/s12094-007-0029-0; PMID: 17403626
- Ohuchida K, Mizumoto K, Lin C, Yamaguchi H, Ohtsuka T, Sato N, et al. MicroRNA-10a is overexpressed in human pancreatic cancer and involved in its invasiveness partially via suppression of the HOXA1 gene. Ann Surg Oncol 2012; 19:2394 - 402; http://dx.doi.org/10.1245/s10434-012-2252-3; PMID: 22407312
- Foley NH, Bray I, Watters KM, Das S, Bryan K, Bernas T, et al. MicroRNAs 10a and 10b are potent inducers of neuroblastoma cell differentiation through targeting of nuclear receptor corepressor 2. Cell Death Differ 2011; 18:1089 - 98; http://dx.doi.org/10.1038/cdd.2010.172; PMID: 21212796
- Varnholt H, Drebber U, Schulze F, Wedemeyer I, Schirmacher P, Dienes HP, et al. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 2008; 47:1223 - 32; http://dx.doi.org/10.1002/hep.22158; PMID: 18307259
- Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, et al. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res 2008; 14:419 - 27; http://dx.doi.org/10.1158/1078-0432.CCR-07-0523; PMID: 18223217
- Vrba L, Garbe JC, Stampfer MR, Futscher BW. Epigenetic regulation of normal human mammary cell type-specific miRNAs. Genome Res 2011; 21:2026 - 37; http://dx.doi.org/10.1101/gr.123935.111; PMID: 21873453
- Li J, Smyth P, Flavin R, Cahill S, Denning K, Aherne S, et al. Comparison of miRNA expression patterns using total RNA extracted from matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC Biotechnol 2007; 7:36; http://dx.doi.org/10.1186/1472-6750-7-36; PMID: 17603869
- Xi Y, Nakajima G, Gavin E, Morris CG, Kudo K, Hayashi K, et al. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 2007; 13:1668 - 74; http://dx.doi.org/10.1261/rna.642907; PMID: 17698639
- Balic M, Pichler M, Strutz J, Heitzer E, Ausch C, Samonigg H, et al. High quality assessment of DNA methylation in archival tissues from colorectal cancer patients using quantitative high-resolution melting analysis. J Mol Diagn 2009; 11:102 - 8; http://dx.doi.org/10.2353/jmoldx.2009.080109; PMID: 19179456
- Harbeck N, Nimmrich I, Hartmann A, Ross JS, Cufer T, Grützmann R, et al. Multicenter study using paraffin-embedded tumor tissue testing PITX2 DNA methylation as a marker for outcome prediction in tamoxifen-treated, node-negative breast cancer patients. J Clin Oncol 2008; 26:5036 - 42; http://dx.doi.org/10.1200/JCO.2007.14.1697; PMID: 18711169
- Bai S, Nasser MW, Wang B, Hsu SH, Datta J, Kutay H, et al. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem 2009; 284:32015 - 27; http://dx.doi.org/10.1074/jbc.M109.016774; PMID: 19726678
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 2009; 37:Database issue D885 - 90; http://dx.doi.org/10.1093/nar/gkn764; PMID: 18940857
- Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res 2006; 16:383 - 93; http://dx.doi.org/10.1101/gr.4410706; PMID: 16449502
- Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 2010; 38:Database issue D204 - 10; http://dx.doi.org/10.1093/nar/gkp1019; PMID: 20015972
- Zhang YJ, Chen CJ, Lee CS, Haghighi B, Yang GY, Wang LW, et al. Aflatoxin B1-DNA adducts and hepatitis B virus antigens in hepatocellular carcinoma and non-tumorous liver tissue. Carcinogenesis 1991; 12:2247 - 52; http://dx.doi.org/10.1093/carcin/12.12.2247; PMID: 1721008