Abstract
Epigenetic modifications, such as aberrant DNA promoter methylation, are frequently observed in cervical cancer. Identification of hypermethylated regions allowing discrimination between normal cervical epithelium and high-grade cervical intraepithelial neoplasia (CIN2/3), or worse, may improve current cervical cancer population-based screening programs. In this study, the DNA methylome of high-grade CIN lesions was studied using genome-wide DNA methylation screening to identify potential biomarkers for early diagnosis of cervical neoplasia. Methylated DNA Immunoprecipitation (MeDIP) combined with DNA microarray was used to compare DNA methylation profiles of epithelial cells derived from high-grade CIN lesions with normal cervical epithelium. Hypermethylated differentially methylated regions (DMRs) were identified. Validation of nine selected DMRs using BSP and MSP in cervical tissue revealed methylation in 63.2–94.7% high-grade CIN and in 59.3–100% cervical carcinomas. QMSP for the two most significant high-grade CIN-specific methylation markers was conducted exploring test performance in a large series of cervical scrapings. Frequency and relative level of methylation were significantly different between normal and cancer samples. Clinical validation of both markers in cervical scrapings from patients with an abnormal cervical smear confirmed that frequency and relative level of methylation were related with increasing severity of the underlying CIN lesion and that ROC analysis was discriminative. These markers represent the COL25A1 and KATNAL2 and their observed increased methylation upon progression could intimate the regulatory role in carcinogenesis. In conclusion, our newly identified hypermethylated DMRs represent specific DNA methylation patterns in high-grade CIN lesions and are candidate biomarkers for early detection.
Introduction
Cervical cancer is one of the most common cancers in women. Early diagnosis is critical in the clinical management of the disease. Development of cervical cancer goes through different premalignant stages, from low-grade cervical intraepithelial neoplasia (CIN1 or LSIL) through high-grade CIN (CIN2/3 or HSIL) to cervical cancer.Citation1 Population-based screening programs for cervical cancer aim to detect CIN2/3 and/or early stage cervical cancer, but the current methodology (Pap smear—cytological screening test to detect potentially pre-cancerous and cancerous cells) is characterized by a significant fraction of false-negative and false-positive results.Citation2,Citation3 Identification of an objective biomarker for detecting women with an increased risk for CIN2/3 or worse (CIN2+) could improve current population-based screening.Citation4
High-risk human papillomavirus (hrHPV) testing identifies more CIN2+ lesions compared with cytology-based cervical screening methods, as has been shown in several population-based studies.Citation5-Citation8 However, especially in younger women, the lower specificity of hrHPV may lead to identification of hrHPV positive women without clinically relevant lesions, resulting in unnecessary referrals to gynecologists and thereby high costs for national health-care systems. To improve the efficacy of population-based screening for cervical cancer, a screening test with a better diagnostic performance (especially higher specificity) needs to be developed.Citation7 Detection of altered DNA methylation might be an ideal tool to improve the current screening methods, since it can be applied on the same specimens used for cytology and/or HPV analyses.Citation9
Abnormal DNA methylation is a well-recognized epigenetic hallmark of cancer cells and has been observed in most malignancies. CpG hypermethylation at the promoters of tumor suppressor genes and other cancer-associated genes is one of the most frequent epigenetic events in malignant cells, and is a common mechanism of tumor suppressor gene inactivation. The list of genes in which promoter CpG islands are hypermethylated in cancer has been continually growing over the last few years. DNA hypermethylation in cervical cancer is a common event with over 50 hypermethylated promoters that have been examined for their methylation status (reviewed by Woodman et al.Citation10). In the past decade, several cervical cancer specific hypermethylated genes have been identified, including C13ORF18, CADM1, CDH1, DAPK1, MAL and TFPI2.Citation11-Citation17 More than 90% of the studies focused on promoter methylation of different genes in cervical cancers, while only few studies examined the relevance of methylation in CIN lesions.Citation11,Citation12,Citation15-Citation17
A number of approaches have been developed to analyze the epigenome on a genome-wide scale (reviewed by LairdCitation18). High-density microarray-based analysis was developed to characterize DNA methylation patterns. Techniques have been applied for genome-wide studies based on bisulfite modification, methylation-sensitive restriction or enrichment of methylated DNA by methyl-binding proteins and a 5′ methyl-cytosine specific antibody. Methyl-DNA immunoprecipitation followed by microarray analysis (MeDIP-chip) is used to enrich for methylated DNA using an antibody and subsequent characterization on CpG Island-Plus-Promoter microarrays.Citation19 Informative genome-wide data reflect methylation differences among different samples, occurring over broad genomic regions.Citation20
In this study, the genome-wide methylation pattern of high-grade CIN lesions was compared with normal cervices using MeDIP-chip to uncover new aberrantly methylated regions in CIN2+ lesions and, thereby, the possibility to develop a high-throughput test for population-based screening. Among the list of differentially methylated regions (DMRs) that we identified, MSP (Methylation Specific PCR) primers were designed for CpG-rich regions. The methylation frequency was tested with these MSP primers on tissue specimens. Finally, our identified potential biomarkers were clinically validated by quantitative MSP (QMSP) on cervical scrapings derived from normal cervices, CIN1–3 and cervical cancer patients.
Results
Identification of hypermethylated DMRs for CIN3 vs. normal cervical epithelium
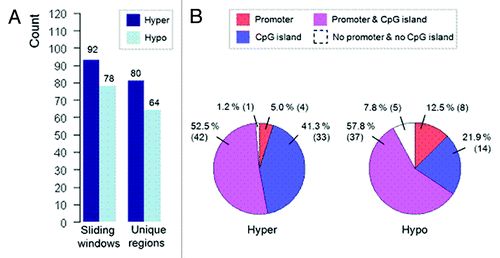
Genome-wide methylation screening (MeDIP-chip) was used to compare DNA methylation profiles of dysplastic cervical cells derived from 15 CIN3 lesions with normal cervical cells derived from 10 patients with a normal cervix to identify specific DMRs, of which the hypermethylated DMRs can be used for clinical validation. Our analysis generated a list of 92 hypermethylated windows corresponding to 80 DMRs (; Table S1) and a list of 78 hypomethylated windows corresponding to 64 DMRs (; Table S2) using a liberal significance cut-off (p < 0.01). These DMRs spanned an average ~500 bps and differently distributed among promoters and non-promoter regions either with CpG or non-CpG islands. The hypermethylated DMRs were represented primarily in promoter-associated CpG islands and non-promoter CpG islands [n = 42 (52.5%) and n = 33 (41.3%), respectively] (), while hypomethylated DMRs showed similar distribution, but were also more pronounced in non-CpG island ().
Figure 1. Distribution of hypermethylated and hypomethylated windows and corresponding DMRs. (A) Number of detected hypermethylated and hypomethylated windows and unique DMRs. (B) Division of sequence context underlying the mapped identified DMRs.
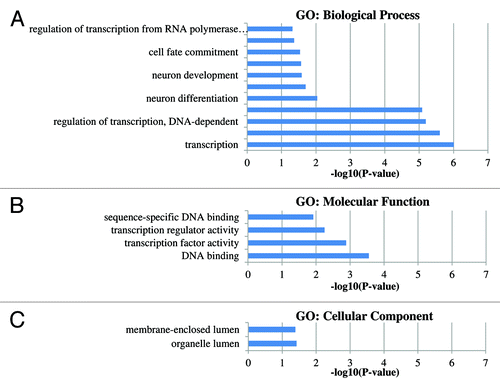
Gene ontology (GO) functional classification was performed to determine if any common functional categories are associated with the genes exhibiting methylation differences between groups. For the gene functional classification and GO analysis, we included all gene-related hypermethylated DMRs with a CpG island (n = 42). The significant GO categories between groups are shown in , while Table S3 lists all GO categories. The most significant GO category determined to be enriched by the analysis was regulation of transcription (GO:0045449, GO:0006355 and GO:0051252) represented by genes like HOXA9, GFI1, CREBZF, ZNF614, etc. (Table S4).
Figure 2. Gene ontology groups displaying the significant GO-terms (p values < 0.05). P values represent the EASE scores given by DAVID. (A) The significant GO-biological process. (B) The significant GO-Molecular function. (C) The significant GO-cellular component.
Validation of DMR specific methylation by MSP
To validate our microarray results, nine hypermethylated DMRs were selected for further analysis using MSP or BSP (Table S5). Validation was restricted to hypermethylated DMRs since these can be easily transferred to clinical diagnostic test using MSP. Only DMRs that covered more than 3 significant windows were selected for validation by MSP. Moreover, DMRs displaying hypomethylation in tumors tend to be more variable between different individuals.Citation21
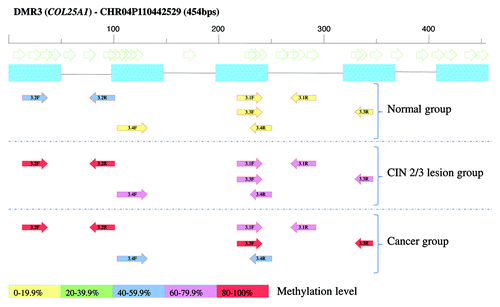
For 8 DMRs, 16 MSP assays were developed to verify and characterize methylation in the same samples that were used for the microarray analysis (13 CIN3 and nine normal cervices, due to insufficient genomic DNA for three samples). An example of one of the MSP assays is displayed in . The marker with the highest significance, a 2.07 kb DMR (chr16:33868738–33870804: DMR9), was analyzed by BSP to identify the most relevant hotspot methylation. BSP analysis showed methylation in CIN3 lesion samples as well as in normal cervical samples used for MeDIP-chip (data not shown). For this reason, and also due to the high number of repetitive regions within DMR9, no suitable MSP could be designed for this DMR and, consequently, quantitative methylation level differences could not be assessed (DMR9 was not further analyzed in this study). In total, 13 out of 16 primer sets covering 8 DMRs were discriminative among CIN3 lesions and normal cervices (Table S6). In CIN3 lesions used for MeDIP-chip analysis hypermethylation was observed in 10–12 out of 13 samples (77–92.3%), for the 13 discriminative primer sets showing the validity of our array results.
Figure 3. Schematic figure of MSP design of the selected hypermethylated DMR3 (COL25A1). Methylation frequency in all samples used for MSP validation is included. Open arrows are representing CpG dinucleotides, blue boxes are probes on Nimblegen array. Colored arrows show the observed methylation frequency of designed primers.
To further validate the discrimination between normal cervical cells and high-grade CIN, additional CIN2/3 lesions, normal cervices and cervical cancers (n = 27) were analyzed. summarizes the MSP results for all samples. Methylation frequency of 13 MSPs showed statistically significant differences between normal cervices and CIN2/3 lesions and between normal cervices and cervical cancers. Analysis of the additional samples revealed that for 8 potential markers, methylation was also frequently observed in normal cervices (44.4–100%) or a low number of cancer samples was methylated (). Therefore, these regions were excluded for the subsequent analysis.
Table 1. Methylation frequency of selected markers in normal, high-grade CIN lesions and cancer tissues
Using the additional samples, we identified 4 marker regions (represented by primer sets DMR1.1, DMR3.3, DMR3.4 and DMR6.1) that displayed low methylation frequencies in normal cervices samples (0–18.5%) and high methylation frequencies in CIN2/3 (73.7–89.5%) and cancer samples (59.3–96.3). These promising candidate markers were further subjected to clinical evaluation on cervical scrapings by QMSP.
Quantitative validation and test performance of selected DMRs as methylation markers by QMSP
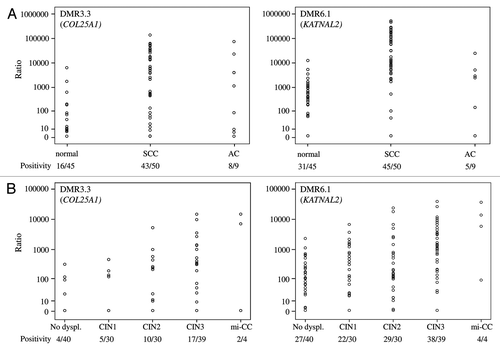
First, the selected DMRs were tested for QMSP conditions. While testing the QMSP conditions on additional normal cervical tissues, relatively high methylation levels were observed for DMR1.1 and DMR3.4 (data not shown). Therefore, these two candidate marker regions were excluded for further analysis. Relative methylation levels of the other two candidate primer sets DMR3.3 (COL25A1) and DMR6.1 (KATNAL2) were determined in scrapings from two cohorts of patients: (1) patients with cancer vs. patients with normal cervices and (2) patients referred with an abnormal smear. In cohort 1, QMSP analysis showed that the frequency and relative level of DNA methylation were significantly different between normal and cancer samples for DMR3.3 (p < 0.001). For DMR6.1, the methylation level differed between the two groups (p < 0.001) (). Age differences might explain variation in methylation levels but, in cohort 1, methylation positivity of the two QMSP assays was not related to age (data not shown). In cohort 2, high relative methylation levels of both DMRs were associated with increasing severity of the underlying lesion (p < 0.001) ().
Figure 4. The relative methylation level of examined DMR3.3 (COL25A1) and DMR6.1 (KATNAL2) on cervical scrapings. Scrapings were derived from women with a normal cervix and cervical cancers (A) and from patients referred with an abnormal smear with different histological diagnosis: CIN1, CIN2, CIN3 and mi-CC (B). The level of methylation for all DMRs increased with the severity of the lesion (all p < 0.001).
In order to determine whether high risk HPV positivity was related to the methylation levels, we compared the HPV status, as determined previously,Citation22 to the methylation levels. This analysis revealed that HPV and methylation levels were not related (data not shown).
To determine whether these candidate regions could be potential biomarkers for diagnostics to distinguish between CIN2+ lesions and no dysplasia/CIN1 lesions, ROC analysis was performed. shows that both markers could significantly distinguish CIN2+ lesions from no dysplasia/CIN1 lesions scraping samples (p < 0.005). In order to compare these new methylation markers to our previously reported ones (JAM3, TERT, EPB41L3Citation22 and C13ORF18Citation11), ROC analysis for these genes were performed on the same patient series to accurately compare diagnostic performance status and showed similar AUCs (). Methylation levels of DMR3.3 (COL25A1) and DMR6.1 (KATNAL2) were higher in the CIN2+ lesions compared with our published methylation markers.
Table 2. ROC analysis of methylation markers
Discussion
In this study, we generated DNA methylomes for normal cervix and CIN3 lesions by combining MeDIP with human CpG island/promoter microarray analysis. Using these DNA methylomes, and by comparing high-grade cervical neoplasia and normal cervices, differentially hypermethylated regions were identified and characterized for their utility for early detection of cervical neoplasia in scrapings. We describe the identification of two new DMRs (COL25A1 and KATNAL2) that show higher quantitative methylation levels as well as higher methylation frequency in CIN2+ lesions than in no dysplasia/CIN1 lesions.
Compared to previous studies,Citation11,Citation16,Citation22,Citation23 these new DMRs strengthen again the increased hypermethylation trend of specific regions during progression of cancer. These DMRs might be used as valuable diagnostic markers in the clinic after population-based validation. Whether these selected methylation biomarkers, either alone or combined with other epidemiological factors and/or genetic markers, could serve as predictors for the risk of progression to cervical cancer should be examined further.
Novak et al. studied genome-wide DNA methylation analyses of isogenic human mammary epithelial cell culture model of transformation and found stepwise DNA methylation changes during immortalization and malignancy.Citation24 Similar data has also been described using methylation specific-multiplex ligation-dependent probe amplification analysis of consecutive stages of HPV16/18-transfected keratinocytes reflecting the carcinogenesis of cervical cancer.Citation25 These studies, together with our own data, illustrate that early alterations in DNA methylation of specific markers may be an important force that drives the significant biological heterogeneity.
Up until now, weCitation11,Citation22 and othersCitation16,Citation26 have identified potential methylation markers to detect cervical cancer and its high-grade precursors with approaches in which normal cervices were compared with cervical cancer tissue specimens. The assumption was that methylation is an early event in the progression of cervical cancer and those methylation markers with a very high sensitivity for cervical cancer would also have a high sensitivity for high-grade CIN lesions. However, we and others found that most markers have a sensitivity of ~70% for high-grade CIN.Citation16,Citation22,Citation23 In the present study, we used high-grade CIN enriched tissue in comparison to normal cervical tissue to generate a DNA methylome specific for high-grade CIN lesions; this strategy resulted in an increased sensitivity to detect high-grade CIN (). Although, these new methylation biomarker candidates showed relatively high clinical sensitivity, the specificity might improve through more detailed analysis of DMRs selected by microarray analyses using in-depth BSP sequencing. Sensitivity and specificity might be further improved by more in-depth analyses of the other 78 DMRs listed in Table S1.
In our present study, we report a list of 80 DMRs obtained by comparing CIN3 lesions vs. normal cervices. Most of the DMRs, and their potentially regulated genes, uncovered through our arrays had not been reported previously as being methylated in high-grade CIN or cervical cancer. Some of the previously described frequently methylated genes found in cervical cancer, were also identified in our study, such as NOL4,Citation27,Citation28 CLUCitation27 and HOXA9.Citation29,Citation30 In addition, genes previously detected to be methylated in other types of cancer were also shown in our DMR list, such as NR2E1,Citation30-Citation33 SALL1,Citation34-Citation36 KCNH1,Citation37 C10ORF26,Citation38 SIM2,Citation39,Citation40 PCDH11X,Citation41,Citation42 EN2,Citation43 TBX15,Citation44,Citation45 HMX2,Citation31,Citation46 FUCA1,Citation47 GFI1Citation32,Citation48,Citation49 and ZNF614,Citation50 indicating that our analysis detected DNA promoter methylation of genes important in carcinogenesis. Nevertheless, our own best-known methylated event in cervical cancer, hypermethylation of the C13ORF18 promoter, was not captured in our array screen. By QMSP we detected methylation of the C13ORF18 promoter in 60% (9 out of 15) of the high-grade CIN specimens used for MeDIP analysis, which is in concordance with our previous report.Citation11 However, methylation frequency in the normal cervices used for the MeDIP analysis was unexpectedly high, resulting in no methylation difference between the two groups (data not shown). Other well-known cervical cancer methylation markers, such as PAX1,Citation26 MAL and CADM1,Citation17 were not included in our list of 80 DMRs either. These genes are known to be methylated in ~70% of high-grade CIN lesions, while most DMRs in our validation analysis showed > 78% methylation in high-grade CIN lesions. Methodologies and tissue source are major variables on studies of methylation frequencies; additionally, DNA quality and quantity in scrapings might be also an important factor. The heterogeneity in specimens and assays may explain why the methylation frequency of certain markers vary widely between studies, as reviewed by Wentzensen et al.Citation9 It is not surprising that we could not find some of the known cervical cancer methylation marker due to differences in populations, specific features of assay protocols, stringency of the statistical analyses or other unidentified factors. Therefore, this may not be indicative of a lack of methylation, but can be explained by relatively small quantitative differences in methylation levels. On the other hand, most DMRs selected for further validation showed increased methylation in high-grade CIN lesions determined by another technique based on bisulfite conversion. This indicates that the identified DMRs have actually increased methylation in high-grade CIN lesions.
Although much of the NimbleGen CpG and promoter microarray covers promoter and the 5′ regions of genes, it is not limited to CpG islands in and around these areas. The CpG array coverage also extends into gene bodies, downstream gene locations, and currently uncharacterized chromosomal regions. The significance of these methylation events, occurring at this type of chromosomal locations and, therefore, with potential effects on gene transcription, is still an open question.Citation51 One of the limitations of the MeDIP-chip approach was that these arrays often include probes that are not within CpG islands or that are made up of repetitive genomic sequences.Citation18 The vast majority of CpG dinucleotides is located within repetitive elements and is methylated in normal tissue. In a typical MeDIP experiment, several classes of repeats are recovered with high efficiency (up to 100%) due to high methylation content of these elements.Citation52 Our top DMR candidate was localized in a repetitive element rich region and we were not able to validate the methylation differences by PCR-based techniques. The presence of bulk quantities of highly methylated repetitive DNA among captured fragments influences the sensitivity of MeDIP-chip based marker discovery. This would be a challenge even for MeDIP-sequencing and encourage the development of a more precise approach.
Understanding the role of tissue-specific differential methylation in the context of non-genic regions, including repetitive sequences that we did not study, will also be critical, especially in light of recent genome-wide association studies of complex diseases that have revealed many putative causative variants to be located within non-genic regions. Future studies will undoubtedly address many of these important questions concerning the role of DNA methylation in genome function.
In addition, the number of identified discriminative hypermethylated and hypomethylated DMRs was ~150 (almost equally divided between the two states), which points to the simultaneous presence of aberrant hypermethylation as well as hypomethylation. This is in line with previously published results on colon cancer.Citation51 The importance of aberrant DNA hypomethylation in cancer is much less known and the role of this phenomenon in cancer development remains poorly understood.Citation53 Loss of methyl-cytosine may result in re-expression of proto-oncogenes or imprinted genes, as well as activation of viral and parasitic transposons, thus contributing to genomic instability. The study of Missaoui et al.Citation54 reported that DNA from invasive squamous cell cancer was more often hypomethylated when compared with DNA from pre-neoplastic lesions and from normal cervix, although there was no significant difference of global DNA methylation content among groups. A similar number of hypomethylated DMRs compared with hypermethylation, as observed in our current study suggests that genome-wide hypomethylation could be an early event in cervical carcinogenesis and may contribute to cancer progression as has been described for hypermethylation by Baylin and Ohm.Citation55 Hypomethylated DMRs might be used as indicative of cancer risk, although further studies are needed to refine this hypothesis.
Finally, one of the main questions in this field arises from the inadequate understanding of the long-term behavior of these lesions: which lesion will become malignant and when? To understand, and to describe a disease, including its prognosis, it is important to know the role of those identified regions/genes in cancer development. Two genes associated with our identified DMRs, COL25A1 and KATNAL2, belonging to the integral membrane function and cytoskeleton, respectively, showed more frequent methylation with increasing severity of the underlying lesion, which is in line with the hypothesis that loss of transcription may lead to a more invasive phenotype of the cell. Promoter DNA methylation has not been described for these two genes before. COL25A1 has been described to be involved in Alzheimer disease,Citation56,Citation57 whereas the function for KATNAL2 is mainly unknown. Validating these two markers as new tools for early detection of high-grade CIN in cervical scrapings in large independent cohorts and investigating their role in normal cervix and during cervical carcinogenesis will be the focus of our future research.
In conclusion, our study is the first genome-wide survey using MeDIP-chip comparing the DNA methylome of high-grade CIN lesions to normal cervices in order to identify potential biomarkers for early detection of high-grade CIN lesions. We present the discovery of a list of novel hypermethylated DMRs in high-grade CIN lesions. QMSP assays developed for two DMRs (COL25A1 and KATNAL2) showed a high rate of methylation in an independent set of high-grade CIN cervical lesions. Identified DMRs could serve as candidate biomarkers, to improve early detection of precancerous lesions, as well as to provide a better insight in cervical carcinogenesis.
Patients and Methods
General strategy
To characterize the DNA methylome of CIN3 lesions and to identify new CIN2+ methylation markers, we applied the following strategy: first, MeDIP with subsequent microarray analysis was performed on DNA isolated from frozen macrodissected epithelial tissue of CIN3 lesions (n = 15) and normal cervices (n = 10). In order to identify putative DMRs, microarray data analysis was performed using a sliding-window approach followed by linear regression. Overlapping significant windows were merged into DMRs and ranked by p-values. In the second step, nine DMRs which covered more than 3 significant windows were selected for validation by MSP on DNA isolated from macrodissected frozen epithelial tissue of cervical cancers (n = 27), CIN2/3 lesions (n = 19) and normal cervices (n = 27). The most significantly differentiating methylation markers (with the least methylation in normal cervices) were selected for further quantitative validation by QMSP on cervical scrapings from a large series of cervical cancer patients (n = 59) and a similar age group of controls (n = 45) to get a first impression of their diagnostic performance. Finally, their potential as a diagnostic tool was evaluated in a large series of scrapings (n = 143) from patients, referred to our department with an abnormal Pap smear, taken during population-based screening.
Patient samples
All patients, referred to our outpatient clinic for (possible) cervical neoplasia, are routinely asked to participate in various studies on biomarkers for cervical neoplasia during their initial visit at the outpatient clinic at the University Medical Centre Groningen (UMCG). Frozen tissue and cervical scrapings for the present study were prospectively collected and stored in our tissue bank from cervical cancer patients, from patients with normal cervices planned to undergo a hysterectomy for non-malignant reasons and from patients referred with an abnormal Pap smear. Indications for hysterectomy were fibroids, prolaps uteri, adenomyosis, hypermenorrhea or a combination of these. All cervical tissues that were used as controls were judged as histopathological normal. All patients referred with an abnormal Pap smear were diagnosed by biopsy or LLETZ (large loop excision of the transformation zone) and all tissue samples were scored by an experienced gynecologic pathologist (H.H.). Clinicopathological data were retrieved from patient files and stored in a large anonymous database. For all cervical cancer patients, an examination under general anesthesia was performed for staging in accordance with the International Federation of Gynecology and Obstetrics (FIGO) criteria. All patients from whom material was obtained gave written informed consent. This study was approved by and followed the ethical guidelines of the Institutional Review Board of the UMCG.
For MeDIP-chip analysis, frozen tissue specimens from 15 CIN3 lesions and 10 normal cervices from our tissue bank were randomly selected. Macrodissection was performed in order to enrich for the dysplastic or normal cervical cells from CIN3 lesions and normal cervices, respectively. Median age of the CIN3 patients was 36 y (range 31–45). Median age of the women with normal cervices was 41 y (range 38–44).
For MSP analysis, additional frozen tissue specimens to obtain in total 19 CIN2/3 lesions, 27 normal cervices and 27 cervical cancers, all were randomly selected. Stage of cervical cancer patients was: 13 (48%) FIGO stage IB, 7 (26%) FIGO stage IIA, 4 (15%) FIGO stage IIB, 1 (4%) FIGO stage IIIB and 2 (7%) FIGO stage IV. Median age of the CIN patients was 34 y (range 30–45), of women with a normal cervix 44 y (range 32–66) and of cervical cancer patients 46 y (range 25–87).
QMSP analysis was first performed on cervical scrapings from our tissue bank from 59 randomly selected cervical cancer patients and 45 normal cervices and subsequently on scrapings from 143 consecutive patients referred with an abnormal Pap smear to our outpatient clinic. The stage of cervical cancer patients was: 30 (51%) FIGO stage IB, 9 (15%) FIGO stage IIA, 14 (24%) FIGO stage IIB, 5 (8%) FIGO stage IIIB and 1 (2%) FIGO stage IV. Histological classification of the cervical cancer patients was: 50 (85%) squamous cell carcinoma and 9 (15%) adenocarcinoma. Patients referred with an abnormal Pap smear were divided in: 40 (28%) without dysplasia (no dysplasia), 30 (21%) CIN1, 30 (21%) CIN2, 39 (27%) CIN3 and 4 (3%) micro-invasive squamous cell cervical carcinoma (miCC). Median age was 45 y (range 27–85) for cervical cancer patients, 48 y (range 30–68) for controls and 35 y (range 20–65) for patients referred with an abnormal Pap smear.
DNA isolation
Genomic DNA was isolated from frozen macrodissected samples and cervical scrapings by overnight Proteinase K treatment, phenol-chloroform extraction, ethanol precipitation and RNase digestion as described previously.Citation22 Genomic DNA was amplified in a multiplex PCR according to the BIOMED-2 protocol, to check the DNA quality.
Methylated DNA immunoprecipitation (MeDIP)
MeDIP assay was performed as described previouslyCitation19 with the following modifications. DNA samples (5 µg) were sheared to a size range of 300–1,000 bps using a Bioruptor™ UCD-200 (Diagenode). Random fragmented DNA was denatured at 95°C for 10 min and snapped-cooled on ice for at least 5 min. Four µg of DNA was immunoprecipitated as described previouslyCitation58 using 50 µl Dynabeads (M-280 sheep anti-mouse IgG, Dynal Biotech).Citation59
Real-Time qPCR on immunoprecipitated samples
Real-Time qPCR was performed for analysis of enrichment. Using an ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems), each qPCR was performed in 10 µl total volume, which contained 5 µl 2X SYBRGreen PCR master mix (Applied Biosystems), 1.5 µl primer mix (150 µM each primer), 250 pg of input DNA or 2 µl of enriched samples. Condition of the reaction was: 95°C 1 min, 95°C 15 sec 60°C 1 min. Each reaction was performed in triplicate. Primer sequences of human H19 and UBE were used as methylated and unmethylated controls, respectively.Citation19 The primer pair 4994 (F 5′ GGGAATATAAGGAGCGCACA 3′ R, 5′ TCGGTTAAAACGGTCAGGTC 3′) was used as an additional positive control to assess the MeDIP-enrichment by qPCR.Citation60 Enrichment was evaluated by calculation of the ratio between input and immunoprecipitated DNA by methylated and unmethylated control sequences and should be at least 15-fold.
Microarray and microarray hybridization
The NimbleGen Human CpG Island-Plus-Promoter array (Nimblegen Systems GmbH) was used to determine the methylation status in the MeDIP-enriched DNA of each sample. The array contains 385,000 oligonucleotide probes covering all UCSC-annotated CpG islands and promoter regions for all RefSeq genes. Each human promoter region covers a region of 1 kb and small CpG islands are extended at both ends for a total coverage of 700 bps (www.nimblegen.com). Fluorescent labeling, hybridization and scanning were performed according to NimbleGen by ImaGenes service (www.imagenes-bio.de). Input samples were labeled with Cy3 and enriched samples were labeled with Cy5, as reported previously.Citation19
Microarray data analysis and annotation
The array signals were expressed as log2 (enriched/input) ratios, are available at http://www.ncbi.nlm.nih.gov/geo/ under accession number GSE36319 and is compliant to the MIAME guidelines. The signal distributions within patient groups were quantile normalized and mean-adjusted across groups (Figs. S1–3). Specific DMRs between normal cervices (n = 10) and CIN3 lesions (n = 15) were identified using a sliding window approach, which was implemented as follows: Let Ykj|i denote the average log2(enriched/input) signal recorded for the jth three-probe window (~500 bps in length) in the ith tiled region (e.g., CpG island or promoter) for individual k. We considered the following linear regression model: Ykj|i = μ0 + βj|igk + εkj|i, where μ0 is the overall signal mean, εkj|i is a normally distributed error term, and gk is an indicator variable taking the values 0 or 1 depending on whether individual k belongs to the normal cervices or the CIN3 lesion group, respectively. We tested the null hypothesis: H0: βj|i = 0 against the alternative HA: βj|i ≠ 0. Hence, significant DMRs for which βj|i > 0 provide evidence for hypermethylation in CIN3 lesions relative to normal cervices, whereas βj|i < 0 provide evidence for hypomethylation. We advanced the three-probe window within the ith tiled region (e.g., CpG island or promoter) one probe at a time, and subsequently jumped to j+1, j+2, j+3,…, n until all tiled regions were visited. Finally, we ranked all tested windows according to their significance level, and selected a liberal subset of regions at p < 0.01 for further validation analysis. This sliding window approach was implemented in the R computing environment.Citation61
RefSeq Gene annotation was used to determine whether candidate DMRs had an overlap with a promoter region of a gene when at least one gene with a transcription start site was located inside the tiled region. For this part of the analysis we used tiled region information, because the arrays were designed for CpG islands and promoters. Sequence and gene annotation data were downloaded from the UCSC genome (version hg18) browser website with the use of the table browser data retrieval tool.Citation62 The DMR annotation analysis was performed in R.Citation61
Gene functional classification and gene ontology (GO) analysis were performed by DAVID analysis tools including the annotated hypermethylated DMRs with a CpG island in CIN3. Conditions for the analysis were set at lowest for the classification stringency and GO categories with P-value smaller than 0.05 were considered statistically significant.Citation63,Citation64
Bisulfite modification and methylation specific PCR (MSP)
Sodium bisulfite treatment of isolated genomic DNA (1 µg/sample) was performed according to the recommendations of the EZ DNA methylation kit (Zymo Research). MSP design and analysis was performed as previously described.Citation22 Methylated primers were designed by the following criteria: presence of two to six CpG dinucleotides especially at the 3′ end of the primer and recognition of methylated CpG dinucleotides of bisulfite modified DNA. The primers should match with the microarray probe sequences, annealing temperature should be at 60°C and the size of the amplicon should be between 70–150 bps to fit for quantitative MSP (QMSP) analysis. Regions of interest were amplified by specific primers, which are summarized in Table S5. Each reaction was performed in 25 µl mix, containing: 500 nM MSP primers, 2 µl of bisulfite treated DNA (approximately 20 ng), standard PCR components (Applied Biosystems) and 0.2 µl 5 U/µl AmpliTaq DNA polymerase (Applied Biosystems). Condition of the MSP was: 95°C for 10 min, 95°C for 1 min, 60°C 50 sec, 72°C 50 sec for 40 cycles, with a final step at 72°C for 7 min. Leukocyte DNA from healthy women was used as negative control and in vitro methylated (by SssI enzyme) leukocyte DNA was used as positive control for each PCR. Conditions of bisulfite sequencing PCR (BSP) was the same as for MSP, only primer design was different. BSP primers are enlisted in Table S5.
Quantitative methylation specific PCR (QMSP)
QMSP was performed as described previously by our groupCitation11Citation22 with an internal dual-labeled hybridization probe (TaqMan®, Applied Biosystems) for quantitative analyses. Primers and probes are summarized in Table S5. β–Actin was used as a methylation independent internal reference gene.Citation11,Citation22 QMSP reactions were performed in 20 µl final volume, containing: 10 µl of 2* QuantiTech Probe Master Mix (Qiagen Benelux), 600 nM of forward and reverse primers (Invitrogen), 250 nM of hybridization probe (Eurogentec) and 5 µl bisulfite modified DNA. Data points were collected during the 50 cycles. Each sample was analyzed in triplicate by ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems). Negative and positive controls were the same as used for MSP. Standard curve analysis was performed on each plate and by each primers-probe set on serial dilutions of positive controls. The relative level of methylation of the region of interest was determined by the following calculation: the average quantity of the methylated region of interest to the average quantity of the reference β–Actin gene and multiplied by 10,000.
Statistical analysis of PCR based methylation assays
Statistical analysis was performed using SPSS software package (SPSS 16.0). Categorical data were analyzed using the χ2 test (with Fisher’s exact test for small numbers when appropriate), while methylation levels were analyzed using the Mann-Whitney U test for two groups or the Kruskal-Wallis test for more than two groups. Associations between positive methylation and age were analyzed with Student T-test. Receiver operating characteristic (ROC) curves were generated to determine specificity and sensitivity for CIN2+ (combination of CIN2/3 and miCC) vs. no dysplasia/CIN1 samples for each QMSP assay. The area under the ROC curve (AUC) was used as a measure of test performance to determine if an assay could discriminate between CIN2+ and no dysplasia/CIN1 samples. P-values lower than 0.05 were considered statistically significant.
| Abbreviations: | ||
| CIN | = | cervical intraepithelial neoplasia |
| hrHPV | = | high-risk human papillomavirus |
| LLETZ | = | large loop excision of the transformation zone |
| FIGO | = | The International Federation of Gynaecology and Obstetrics |
| miCC | = | micro-invasive squamous cell cervical carcinoma |
| MeDIP | = | methylated DNA immunoprecipitation |
| Real-Time qPCR | = | Real-Time quantitative PCR |
| BSP | = | bisulfite sequencing PCR |
| MSP | = | methylation specific PCR |
| QMSP | = | quantitative methylation specific PCR |
| DMR | = | differentially methylated region |
| ROC | = | receiver operating characteristic |
| AUC | = | area under curve |
| CGI | = | CpG island |
| GO | = | gene ontology |
Additional material
Download Zip (117.3 KB)Acknowledgments
This study was supported by the Dutch Cancer Society (NKB) (project-number RUG 2004-3161) and by OncoMethylome Sciences S.A. (currently known as MDxHealth S.A.), Liège, Belgium. CG was funded by the German Ministry of Education and Research (BMBF) within the NGFN-PLUS project PKT-01GS08111. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript
Disclosure of Potential Conflicts of Interest
Prof A.G.J. van der Zee was member of the scientific advisory board of OncoMethylome Sciences S.A. (currently known as MDxHealth S.A.), Liège, Belgium.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/22301
Related Research Data
References
- Wentzensen N, Klug SJ. Early detection of cervical carcinomas: finding an overall approach. Dtsch Arztebl Int 2008; 105:617 - 22; PMID: 19471627
- Robertson JH, Woodend B. Negative cytology preceding cervical cancer: causes and prevention. J Clin Pathol 1993; 46:700 - 2; http://dx.doi.org/10.1136/jcp.46.8.700; PMID: 8408692
- Koss LG. Cervical (Pap) smear. New directions. Cancer 1993; 71:Suppl 1406 - 12; http://dx.doi.org/10.1002/cncr.2820710405; PMID: 8381706
- Arbyn M, Ronco G, Cuzick J, Wentzensen N, Castle PE. How to evaluate emerging technologies in cervical cancer screening?. Int J Cancer 2009; 125:2489 - 96; http://dx.doi.org/10.1002/ijc.24774; PMID: 19626591
- Bulkmans NW, Rozendaal L, Snijders PJ, Voorhorst FJ, Boeke AJ, Zandwijken GR, et al. POBASCAM, a population-based randomized controlled trial for implementation of high-risk HPV testing in cervical screening: design, methods and baseline data of 44,102 women. Int J Cancer 2004; 110:94 - 101; http://dx.doi.org/10.1002/ijc.20076; PMID: 15054873
- Mayrand MH, Duarte-Franco E, Rodrigues I, Walter SD, Hanley J, Ferenczy A, et al, Canadian Cervical Cancer Screening Trial Study Group. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. N Engl J Med 2007; 357:1579 - 88; http://dx.doi.org/10.1056/NEJMoa071430; PMID: 17942871
- Ronco G, Giorgi-Rossi P, Carozzi F, Confortini M, Dalla Palma P, Del Mistro A, et al, New Technologies for Cervical Cancer Screening Working Group. Results at recruitment from a randomized controlled trial comparing human papillomavirus testing alone with conventional cytology as the primary cervical cancer screening test. J Natl Cancer Inst 2008; 100:492 - 501; http://dx.doi.org/10.1093/jnci/djn065; PMID: 18364502
- Naucler P, Ryd W, Törnberg S, Strand A, Wadell G, Elfgren K, et al. Efficacy of HPV DNA testing with cytology triage and/or repeat HPV DNA testing in primary cervical cancer screening. J Natl Cancer Inst 2009; 101:88 - 99; http://dx.doi.org/10.1093/jnci/djn444; PMID: 19141778
- Wentzensen N, Sherman ME, Schiffman M, Wang SS. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol Oncol 2009; 112:293 - 9; http://dx.doi.org/10.1016/j.ygyno.2008.10.012; PMID: 19054549
- Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer 2007; 7:11 - 22; http://dx.doi.org/10.1038/nrc2050; PMID: 17186016
- Yang N, Eijsink JJ, Lendvai A, Volders HH, Klip H, Buikema HJ, et al. Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer Epidemiol Biomarkers Prev 2009; 18:3000 - 7; http://dx.doi.org/10.1158/1055-9965.EPI-09-0405; PMID: 19843677
- Feng Q, Balasubramanian A, Hawes SE, Toure P, Sow PS, Dem A, et al. Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst 2005; 97:273 - 82; http://dx.doi.org/10.1093/jnci/dji041; PMID: 15713962
- Lim EH, Ng SL, Li JL, Chang AR, Ng J, Ilancheran A, et al. Cervical dysplasia: assessing methylation status (Methylight) of CCNA1, DAPK1, HS3ST2, PAX1 and TFPI2 to improve diagnostic accuracy. Gynecol Oncol 2010; 119:225 - 31; http://dx.doi.org/10.1016/j.ygyno.2010.07.028; PMID: 20708786
- Missaoui N, Hmissa S, Trabelsi A, Traoré C, Mokni M, Dante R, et al. Promoter hypermethylation of CDH13, DAPK1 and TWIST1 genes in precancerous and cancerous lesions of the uterine cervix. Pathol Res Pract 2011; 207:37 - 42; http://dx.doi.org/10.1016/j.prp.2010.11.001; PMID: 21129853
- Steenbergen RD, Kramer D, Braakhuis BJ, Stern PL, Verheijen RH, Meijer CJ, et al. TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J Natl Cancer Inst 2004; 96:294 - 305; http://dx.doi.org/10.1093/jnci/djh031; PMID: 14970278
- Overmeer RM, Louwers JA, Meijer CJ, van Kemenade FJ, Hesselink AT, Daalmeijer NF, et al. Combined CADM1 and MAL promoter methylation analysis to detect (pre-)malignant cervical lesions in high-risk HPV-positive women. Int J Cancer 2011; 129:2218 - 25; http://dx.doi.org/10.1002/ijc.25890; PMID: 21190187
- Hesselink AT, Heideman DA, Steenbergen RD, Coupé VM, Overmeer RM, Rijkaart D, et al. Combined promoter methylation analysis of CADM1 and MAL: an objective triage tool for high-risk human papillomavirus DNA-positive women. Clin Cancer Res 2011; 17:2459 - 65; http://dx.doi.org/10.1158/1078-0432.CCR-10-2548; PMID: 21389098
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 2010; 11:191 - 203; http://dx.doi.org/10.1038/nrg2732; PMID: 20125086
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853 - 62; http://dx.doi.org/10.1038/ng1598; PMID: 16007088
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 66; http://dx.doi.org/10.1038/ng1990; PMID: 17334365
- Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012; 22:271 - 82; http://dx.doi.org/10.1101/gr.117523.110; PMID: 21659424
- Eijsink JJ, Lendvai A, Deregowski V, Klip HG, Verpooten G, Dehaspe L, et al. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J Cancer 2012; 130:1861 - 9; http://dx.doi.org/10.1002/ijc.26326; PMID: 21796628
- Yang N, Nijhuis ER, Volders HH, Eijsink JJ, Lendvai A, Zhang B, et al. Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cell Oncol 2010; 32:131 - 43; PMID: 20208141
- Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res 2009; 69:5251 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-4977; PMID: 19509227
- Henken FE, Wilting SM, Overmeer RM, van Rietschoten JG, Nygren AO, Errami A, et al. Sequential gene promoter methylation during HPV-induced cervical carcinogenesis. Br J Cancer 2007; 97:1457 - 64; http://dx.doi.org/10.1038/sj.bjc.6604055; PMID: 17971771
- Lai HC, Lin YW, Huang TH, Yan P, Huang RL, Wang HC, et al. Identification of novel DNA methylation markers in cervical cancer. Int J Cancer 2008; 123:161 - 7; http://dx.doi.org/10.1002/ijc.23519; PMID: 18398837
- Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res 2008; 68:2661 - 70; http://dx.doi.org/10.1158/0008-5472.CAN-07-5913; PMID: 18413733
- Wang SS, Smiraglia DJ, Wu YZ, Ghosh S, Rader JS, Cho KR, et al. Identification of novel methylation markers in cervical cancer using restriction landmark genomic scanning. Cancer Res 2008; 68:2489 - 97; http://dx.doi.org/10.1158/0008-5472.CAN-07-3194; PMID: 18381458
- Apostolidou S, Hadwin R, Burnell M, Jones A, Baff D, Pyndiah N, et al. DNA methylation analysis in liquid-based cytology for cervical cancer screening. Int J Cancer 2009; 125:2995 - 3002; http://dx.doi.org/10.1002/ijc.24745; PMID: 19609949
- Rauch TA, Wang Z, Wu X, Kernstine KH, Riggs AD, Pfeifer GP. DNA methylation biomarkers for lung cancer. Tumour Biol 2012; 33:287 - 96; http://dx.doi.org/10.1007/s13277-011-0282-2; PMID: 22143938
- Dunwell T, Hesson L, Rauch TA, Wang L, Clark RE, Dallol A, et al. A genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol Cancer 2010; 9:44; http://dx.doi.org/10.1186/1476-4598-9-44; PMID: 20184741
- Tommasi S, Karm DL, Wu X, Yen Y, Pfeifer GP. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res 2009; 11:R14; http://dx.doi.org/10.1186/bcr2233; PMID: 19250546
- Rauch TA, Zhong X, Wu X, Wang M, Kernstine KH, Wang Z, et al. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci U S A 2008; 105:252 - 7; http://dx.doi.org/10.1073/pnas.0710735105; PMID: 18162535
- Hill VK, Hesson LB, Dansranjavin T, Dallol A, Bieche I, Vacher S, et al. Identification of 5 novel genes methylated in breast and other epithelial cancers. Mol Cancer 2010; 9:51; http://dx.doi.org/10.1186/1476-4598-9-51; PMID: 20205715
- Kuang SQ, Tong WG, Yang H, Lin W, Lee MK, Fang ZH, et al. Genome-wide identification of aberrantly methylated promoter associated CpG islands in acute lymphocytic leukemia. Leukemia 2008; 22:1529 - 38; http://dx.doi.org/10.1038/leu.2008.130; PMID: 18528427
- Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, et al. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics 2010; 5:499 - 508; http://dx.doi.org/10.4161/epi.5.6.12179; PMID: 20484983
- Fassbender A, Lewin J, König T, Rujan T, Pelet C, Lesche R, et al. Quantitative DNA methylation profiling on a high-density oligonucleotide microarray. Methods Mol Biol 2010; 576:155 - 70; http://dx.doi.org/10.1007/978-1-59745-545-9_9; PMID: 19882262
- Cheong HS, Lee HC, Park BL, Kim H, Jang MJ, Han YM, et al. Epigenetic modification of retinoic acid-treated human embryonic stem cells. BMB Rep 2010; 43:830 - 5; http://dx.doi.org/10.5483/BMBRep.2010.43.12.830; PMID: 21189161
- Mishra DK, Chen Z, Wu Y, Sarkissyan M, Koeffler HP, Vadgama JV. Global methylation pattern of genes in androgen-sensitive and androgen-independent prostate cancer cells. Mol Cancer Ther 2010; 9:33 - 45; http://dx.doi.org/10.1158/1535-7163.MCT-09-0486; PMID: 20053773
- Old RW, Crea F, Puszyk W, Hultén MA. Candidate epigenetic biomarkers for non-invasive prenatal diagnosis of Down syndrome. Reprod Biomed Online 2007; 15:227 - 35; http://dx.doi.org/10.1016/S1472-6483(10)60713-4; PMID: 17697502
- Maekawa R, Yagi S, Ohgane J, Yamagata Y, Asada H, Tamura I, et al. Disease-dependent differently methylated regions (D-DMRs) of DNA are enriched on the X chromosome in uterine leiomyoma. J Reprod Dev 2011; 57:604 - 12; http://dx.doi.org/10.1262/jrd.11-035A; PMID: 21685710
- Lopes AM, Ross N, Close J, Dagnall A, Amorim A, Crow TJ. Inactivation status of PCDH11X: sexual dimorphisms in gene expression levels in brain. Hum Genet 2006; 119:267 - 75; http://dx.doi.org/10.1007/s00439-006-0134-0; PMID: 16425037
- Bennett LB, Schnabel JL, Kelchen JM, Taylor KH, Guo J, Arthur GL, et al. DNA hypermethylation accompanied by transcriptional repression in follicular lymphoma. Genes Chromosomes Cancer 2009; 48:828 - 41; http://dx.doi.org/10.1002/gcc.20687; PMID: 19530241
- Chelbi ST, Doridot L, Mondon F, Dussour C, Rebourcet R, Busato F, et al. Combination of promoter hypomethylation and PDX1 overexpression leads to TBX15 decrease in vascular IUGR placentas. Epigenetics 2011; 6:247 - 55; http://dx.doi.org/10.4161/epi.6.2.13791; PMID: 20962579
- Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet 2010; 70:277 - 308; http://dx.doi.org/10.1016/B978-0-12-380866-0.60010-1; PMID: 20920752
- Jin B, Yao B, Li JL, Fields CR, Delmas AL, Liu C, et al. DNMT1 and DNMT3B modulate distinct polycomb-mediated histone modifications in colon cancer. Cancer Res 2009; 69:7412 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-09-0116; PMID: 19723660
- Halaban R, Krauthammer M, Pelizzola M, Cheng E, Kovacs D, Sznol M, et al. Integrative analysis of epigenetic modulation in melanoma cell response to decitabine: clinical implications. PLoS One 2009; 4:e4563; http://dx.doi.org/10.1371/journal.pone.0004563; PMID: 19234609
- Shih IeM, Chen L, Wang CC, Gu J, Davidson B, Cope L, et al. Distinct DNA methylation profiles in ovarian serous neoplasms and their implications in ovarian carcinogenesis. Am J Obstet Gynecol 2010; 203:584 - 22, e1-22; http://dx.doi.org/10.1016/j.ajog.2010.08.003; PMID: 20965493
- Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell 2007; 27:562 - 72; http://dx.doi.org/10.1016/j.molcel.2007.06.039; PMID: 17707228
- Papageorgio C, Harrison R, Rahmatpanah FB, Taylor K, Davis W, Caldwell CW. Algorithmic discovery of methylation “hot spots” in DNA from lymphoma patients. Cancer Inform 2008; 6:449 - 53; PMID: 19259422
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 2010; 52:232 - 6; http://dx.doi.org/10.1016/j.ymeth.2010.06.012; PMID: 20542119
- Wild L, Flanagan JM. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim Biophys Acta 2010; 1806:50 - 7; PMID: 20398739
- Missaoui N, Hmissa S, Dante R, Frappart L. Global DNA methylation in precancerous and cancerous lesions of the uterine cervix. Asian Pac J Cancer Prev 2010; 11:1741 - 4; PMID: 21338225
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction?. Nat Rev Cancer 2006; 6:107 - 16; http://dx.doi.org/10.1038/nrc1799; PMID: 16491070
- Söderberg L, Dahlqvist C, Kakuyama H, Thyberg J, Ito A, Winblad B, et al. Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. FEBS J 2005; 272:2231 - 6; http://dx.doi.org/10.1111/j.1742-4658.2005.04647.x; PMID: 15853808
- Tong Y, Xu Y, Scearce-Levie K, Ptácek LJ, Fu YH. COL25A1 triggers and promotes Alzheimer’s disease-like pathology in vivo. Neurogenetics 2010; 11:41 - 52; http://dx.doi.org/10.1007/s10048-009-0201-5; PMID: 19548013
- Chavez L, Jozefczuk J, Grimm C, Dietrich J, Timmermann B, Lehrach H, et al. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res 2010; 20:1441 - 50; http://dx.doi.org/10.1101/gr.110114.110; PMID: 20802089
- Grimm C, Adjaye J. Analysis of the methylome of human embryonic stem cells employing methylated DNA immunoprecipitation coupled to next-generation sequencing. Methods Mol Biol 2012; 873:281 - 95; http://dx.doi.org/10.1007/978-1-61779-794-1_19; PMID: 22528363
- Butcher LM, Beck S. AutoMeDIP-seq: a high-throughput, whole genome, DNA methylation assay. Methods 2010; 52:223 - 31; http://dx.doi.org/10.1016/j.ymeth.2010.04.003; PMID: 20385236
- Team RDC. A language and environment for statistical computing. 2011, Vienna, Austria.
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 2004; 32:Database issue D493 - 6; http://dx.doi.org/10.1093/nar/gkh103; PMID: 14681465
- Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1 - 13; http://dx.doi.org/10.1093/nar/gkn923; PMID: 19033363
- Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44 - 57; http://dx.doi.org/10.1038/nprot.2008.211; PMID: 19131956