Abstract
The interplay of metabolism and epigenetic regulatory mechanisms has become a focal point for a better understanding of cancer development and progression. In this study, we have acquired data supporting previous observations that demonstrate glutamine metabolism affects histone modifications in human breast cancer cell lines. Treatment of non-invasive epithelial (T-47D and MDA-MB-361) and invasive mesenchymal (MDA-MB-231 and Hs-578T) breast cancer cell lines with the glutaminase inhibitor, Compound 968, resulted in cytotoxicity in all cell lines, with the greatest effect being observed in MDA-MB-231 breast cancer cells. Compound 968-treatment induced significant downregulation of 20 critical cancer-related genes, the majority of which are anti-apoptotic and/or promote metastasis, including AKT, BCL2, BCL2L1, CCND1, CDKN3, ERBB2, ETS1, E2F1, JUN, KITLG, MYB, and MYC. Histone H3K4me3, a mark of transcriptional activation, was reduced at the promoters of all but one of these critical cancer genes. The decrease in histone H3K4me3 at global and gene-specific levels correlated with reduced expression of SETD1 and ASH2L, genes encoding the histone H3K4 methyltransferase complex. Further, the expression of other epigenetic regulatory genes, known to be downregulated during apoptosis (e.g., DNMT1, DNMT3B, SETD1 and SIRT1), was also downregulated by Compound 968. These changes in gene expression and histone modifications were accompanied by the activation of apoptosis, and decreased invasiveness and resistance of MDA-MB-231 cells to chemotherapeutic drug doxorubicin. The results of this study provide evidence to a link between cytotoxicity caused by inhibiting glutamine metabolism with alterations of the epigenome of breast cancer cells and suggest that modification of intracellular metabolism may enhance the efficiency of epigenetic therapy.
Introduction
Accumulated evidence in recent years indicates an existence of an intimate link between the metabolic status and epigenetic regulation of cells.Citation1-Citation4 This is evident by the fact that a variety of small molecules derived from intermediary intercellular metabolism, including S-adenosylmethionine, pyruvate, flavin adenine dinucleotide, nicotine adenine dinucleotide, folate, acetyl coenzyme A, α-ketoglutarate and iron, are essential for proper maintenance of the cellular epigenome.Citation2,Citation3,Citation5 The metabolic status and epigenetic regulation are well-balanced in normal cells, but they are profoundly disturbed in cancer cells, including breast cancer cells.Citation6-Citation8 In a previous study using a panel of human breast cancer cell lines, we demonstrated that metabolite levels, metabolically sensitive epigenetic histone marks and the expression of metabolic and histone modifying enzyme genes are associated with different stages of breast carcinogenesis.Citation9 For example, highly invasive and drug-resistant MDA-MB-231 and Hs-578T mesenchymal breast cancer cells were characterized by altered glutamine-glutamate levels indicative of increased glutamine metabolism, a greater expression of glutaminase (GLS1) and histone deacetylase 9 (HDAC9) genes and a lower level of histone H4 lysine 20 trimethylation and H4 lysine 16 acetylation (H4K16ac) as compared with non-invasive epithelial T-47D and MDA-MB-361 breast cancer cells.Citation9
The results of recent studies have suggested that glutamate is a key player in tumorigenesis,Citation10-Citation12 including breast cancer development and progression.Citation11,Citation13,Citation14 Specifically, Wang et al.Citation13 demonstrated that an aberrant increase in glutaminase-catalyzed hydrolysis of L-glutamine to L-glutamate contributes to tumorigenesis, and Asiago et al.Citation14 reported that an elevated level of glutamate is associated with disease outcome in breast cancer patients; however, the mechanism by which the aberrant glutamine-glutamate metabolic pathway may influence breast cancer tumorigenesis has not been elucidated.
One possible mechanism is associated with the fact that metabolites involved in intracellular glucose and glutamine metabolic pathways are substrates for several histone modifying enzymes, including histone acetyltransferases (HATs) and histone demethylases (HDMs). Specifically, acetyl-CoA is required for the proper functioning of HATs, and 2-oxoglutarate is a co-factor for HDMs; affecting the levels of these substrates influences histone modifications, gene expression and cellular phenotype. For example, inhibiting the production of acetyl-CoA by siRNA silencing of ATP-citrate lyase, reduces global and gene-specific levels of histone acetylation and influences gene expression and cellular phenotype.Citation15 Further, we have demonstrated that low levels of H4K16ac in human MDA-MB-231 breast cancer cells can be partially reversed by using compound 968, a dibenzophenanthridine, to block glutamate production via highly specific inhibition of glutaminase,Citation16 and that the compound 968 treatment significantly reduces the global level of histone H3 lysine 4 trimethylation (H3K4me3).Citation9 Therefore, it is plausible that glutamate production influences histone modifications to reprogram cells.
In this study, we investigated whether metabolic targeting of the epigenome using compound 968 influences gene expression and affects the cellular phenotype. We found that compound 968 treatment substantially reduces cell survival of T-47D, MDA-MB-361, Hs-578T and, especially, MDA-MB-231 human breast cancer cells. The activation of apoptosis, decreased survival and invasiveness of MDA-MB-231 cancer cells, and increased drug sensitivity was accompanied by downregulation of gene expression and reduced histone H3K4me3 at the promoters of oncogenes and tumor suppressor genes, particularly at genes playing a fundamental role in apoptosis, cell cycle, cell adhesion and metastasis.
Results
Effect of compound 968 treatment on human breast cancer cells
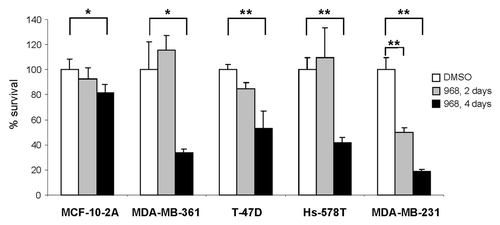
A previous report by Wang et al.Citation13,Citation16 showed that blocking glutamine metabolism via GLS1 inhibition in human breast cancer cells with compound 968 decreases proliferation of MDA-MB-231 cells and hampers tumor formation and progression in vitro and in vivo. Indeed, shows that repeat-dose treatment of MDA-MB-361, T-47D, Hs-578T and MDA-MB-231 breast cancer cell lines with compound 968 was cytotoxic for all studied cancer cells (p < 0.05), with the greatest cytotoxic effect being found in MDA-MB-231 cancer cells (p < 0.01 for both acute single dose and repeat-dose treatments). In contrast, compound 968 had a less pronounced cytotoxic effect on normal breast epithelial MCF-10A cells.
Figure 1. Compound 968 is cytotoxic in multiple human breast cancer cell lines. Cell survival was determined as described in “Materials and Methods” in MCF-10A, MDA-MB-361, T-47D, Hs-578T, and MDA-MB-231 breast cancer cells following 2 and 4 d treatments with 10 μM compound 968 or DMSO control (n = 3). Percent survival was determined by dividing the number of remaining 968 treated cells by the number of remaining viable DMSO treated cells. Assays were performed in triplicate and error bars represent the standard deviation between replicate independent experiments. Asterisks indicate a significant difference from DMSO control; * p < 0.05; ** p < 0.01.
Compound 968 alters the expression of cancer-related genes in MDA-MB-231 cancer cells
shows that treatment of MDA-MB-231 cells with Compound 968 resulted in the downregulation of 20 genes (fold change ≥ 2.0 and p < 0.05), whereas expression of 52 genes (62%) showed either no difference, or changed below the ≥ 2.0 fold cut off values (Table S1). Among these genes, several genes, including critical VHL, MGMT and FHIT tumor suppressor genes, were upregulated 1.6-, 1.8- and 1.9-fold, respectively. Additionally, 12 genes displayed Ct values > 33 cycles, suggesting that these genes are not expressed in MDA-MB-231 cells (Table S1). The majority (75%) of the genes downregulated by Compound 968 treatment are involved in the regulation of apoptosis, including 11 anti-apoptotic genes, e.g., AKT, BCL2, BCL2L1, CCND1, CDKN3, ERBB2, ETS1, JUN, KITLG, MYB and MYC, which were downregulated 2.0- to 5.5-fold; 2 pro-apoptotic genes, e.g., RASSF1 and RB1, which were downregulated 4.1- and 2.4-fold; and, two genes that possess anti-apoptotic and pro-apoptotic functions, e.g., E2F1 and TP73, that were downregulated 3.4- and 3.8-fold, respectively (). Treatment of MDA-MB-231 cells with compound 968 resulted also in a decreased expression of 11 genes involved in cell adhesion, migration, and invasion ().
Figure 2. Compound 968-induced downregulation of gene expression correlates with reduced histone H3K4me3 at corresponding gene promoters. (A) Differentially expressed genes (fold change ≥ 2.0 and p < 0.05; n = 3) in MDA-MB-231 cancer cells in response to 2 d treatment with 10 μM compound 968 (black bars) or DMSO control (white bars) are shown. The data are presented as an average fold-change in the expression of each gene in the Compound 968-treated cells relative to expression in the untreated control cells, which were assigned the value 1. Fold-changes were determined by normalizing expression to B2-microglobulin and RPL13A. Error bars = SD. Relative changes in histone H4K14ac (B) and histone H3K4me3 (C) at the promoters of differentially expressed genes. A ChIP assay was performed with primary antibodies against acetyl-histone H4 lysine 16 (H4K16ac), trimethyl-histone H3 lysine 4 (H3K4me3), or rabbit IgG. Purified DNA from immunoprecipitates and from input DNA was analyzed by qPCR with the Human Oncogenes and Tumor Suppressor Genes EpiTect ChIP PCR Arrays (Qiagen). The data are presented as fold-change in Compound 968-treated MDA-MB-231 cells relative to the DMSO control MDA-MB-231 cells after normalization to input DNA and control IgG. Error bars = SD. Asterisks denote a difference (fold change ≥ 1.5) from the untreated control cells.
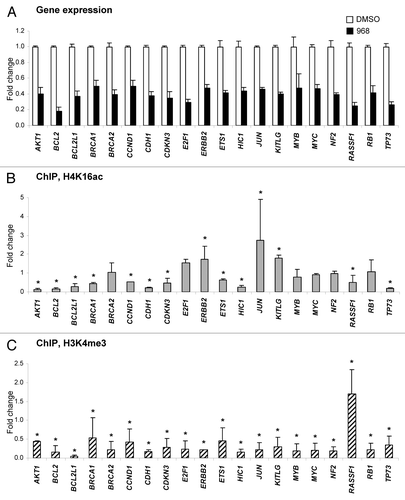
Table 1. Differentially expressed genes in MDA-MB-231 breast cancer cells treated with compound 968 (fold change ≥ 2.0 and p < 0.05; n = 3)
Compound 968 affects the gene-specific histone H4K16ac and H3K4me3 patterns in MDA-MB-231 cancer cells
Next, we determined if the gene expression changes induced by Compound 968 are associated with gene-specific changes in the histone modification pattern at the promoters of the same tumor suppressor genes and oncogenes. shows that among the genes that displayed significant gene expression changes only three genes exhibited an increase in histone H4K16ac at their promoters, whereas in six genes the level histone H4K16ac did not change and in 11 other genes histone H4K16 significantly decreased (fold change ≥ 1.5). In contrast, as expected, in 19 out of 20 genes, we observed a decrease in the gene-specific histone H3K4me3 level () similar to what was found previously at the global level.Citation9
Compound 968 alters the expression of DNA and histone modifying genes in MDA-MB-231 cancer cells
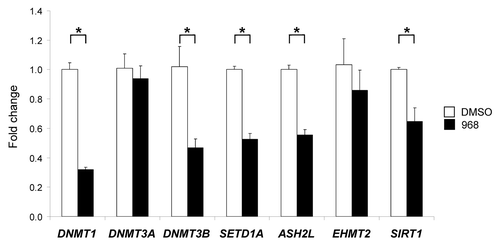
To evaluate further the effect of compound 968 treatment on the functioning of the DNA and histone methylation machinery, qRT-PCR was conducted to examine the expression of DNA methyltransferases (DNMTs) DNMT1, DNMT3A and DNMT3B, histone methyltransferases (HMTs), including, histone H3K4 methyltransferases SETD1A and ASH2L,Citation17,Citation18 histone H3K9 methyltransferase EHMT2Citation19 and histone H4K16 deacetylase SIRT1.Citation20
shows that treatment of MDA-MB-231 cells with compound 968 resulted in the significant downregulation of DNMT1, DNMT3B, SETD1A, ASH2L and SIRT (p < 0.05). In contrast, expression of DNMT3A and EHMT2 was not affected by the compound 968 treatment.
Figure 3. Expression of DNMT1, DNMT3A, DNMT3B, SETD1A, ASH2L, EHMT2 and SIRT1 histone-modifying genes in the DMSO control and compound 968-treated MDA-MB-231 human breast cancer cells. Total RNA from the untreated and compound 968-treated cells was used to evaluate transcript abundance of DNMT1, DNMT3A, DNMT3B, SETD1A, ASH2L, EHMT2 and SIRT1 genes. The gene expression was determined by qRT-PCR as detailed in “Materials and Methods.” Data are presented as an average fold change in the expression of each gene in the Compound 968-treated cells relative to that in the corresponding DMSO control cells, which were assigned a value 1. Asterisks denote a significant (p < 0.05) difference from the DMSO control cells (n = 3)
Compound 968 activates apoptosis, decreases invasiveness, and increases drug sensitivity in MDA-MB-231 cancer cells
Finally, we studied whether compound 968-mediated alterations in gene expression and histone modifications in MDA-MB-231 cancer cells affects their key phenotypic features, such as evasion to apoptosis, invasiveness, and resistance to chemotherapeutic drugs. shows that the activity of effector caspases 3/7 was 1.7-times greater in compound 968-treated MDA-MB-231 cells than in untreated cells. Additionally, the expression of initiator caspase 8 was significantly upregulated also (Fig. S1).
Figure 4. Compound 968 activates apoptosis, decreases invasiveness, and increases drug sensitivity in MDA-MB-231 cancer cells. (A) Activity of caspase 3/7 in MDA-MB-231 cells treated with DMSO (white bar) or compound 968 (black bar). The data are presented as percent change from DMSO-treated cells. (B) Cell invasion activity of MDA-MB-231 cells treated with DMSO (white bar) or compound 968 (black bar). The data are presented as percent change of migrating MDA-MB-231 cells treated with Compound 968 to the lower surface of the membrane from DMSO-treated cells. (C) Sensitivity of MDA-MB-231 cells treated with DMSO (white bar) or compound 968 (black bar) to doxorubicin treatment. The data are presented as mean ± SD (n = 3). Asterisks indicate a significant difference from DMSO-treated cells.
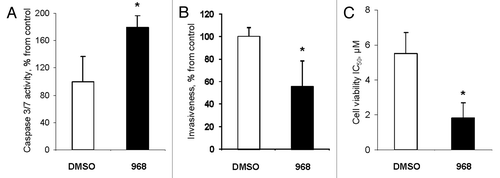
Treatment of MDA-MB-231 breast cancer cells with Compound 968 substantially reduced the invasiveness of cells (). Additionally, compound 969-treated MDA-MB-231 cells exhibited an increased sensitivity to doxorubicin (DOX), a drug that is commonly used to treat breast cancer. This was evidenced by the fact that compound 968 strongly lowered IC50 for DOX from 5.5 μM to 1.8 μM, or 67% in MDA-MB-231 cells ().
Discussion
Emerging evidence has indicated that targeted blocking of glutaminase and glutamine metabolism in cancer cells via either small molecules, e.g., compound 968 and bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), or siRNACitation21 may improve disease outcome in cancer, including breast cancer.Citation11,Citation13,Citation22 In the present study we demonstrate that treatment of human breast cancer MDA-MB-231 cells with compound 968 reduces cancer cell survival, which was accompanied by downregulation of critical cancer-related genes closely associated with breast cancer progression, particularly at genes playing a fundamental role in apoptosis, cell cycle, and cell adhesion and metastasis. The majority of the downregulated genes are involved in the regulation of apoptosis (). Importantly, 11 of the downregulated apoptotic pathway genes were anti-apoptotic oncogenes, a number substantially greater than the two downregulated pro-apoptotic tumor suppressor genes. This may shift the balance between apoptotic and anti-apoptotic processes in MDA-MB-231 cells toward activation of apoptosis, with a resultant increase in the cytotoxic effects of compound 968. This suggestion is supported by wealth of data showing that inhibition of several anti-apoptotic oncogenes, which were found to be downregulated by compound 968 treatment, including AKT1, BCL2, CCND1, ERBB2, ETS1, JUN and MYC, induces apoptosis in cancer cells of different cancer types, including breast cancer cells.Citation23-Citation30 Furthermore, there is convincing evidence that the loss of the RB1 gene, one of the tumor-suppressor genes downregulated by compound 968 treatment, may trigger unscheduled apoptosis.Citation31,Citation32
Treatment of MDA-MB-231 cells with compound 968 also resulted in a decreased expression of 11 genes involved in cell migration, invasion and metastasis (). Previously, we demonstrated that reversing the highly metastatic mesenchymal phenotype of MDA-MB-231 cancer cells increases their sensitivity to chemotherapeutic agents via activation of apoptotic program.Citation33 Similarly, several studies have shown anti-oncogenic effects by suppressing the expression of invasion and metastasis genes, including AKT1, CCND1, E2F1, ERRB2 and JUN.Citation27,Citation34,Citation35 This suggests that the compound 968 cytotoxicity may be also attributed to downregulation of genes that promote invasion and metastasis. Importantly, the observed changes in gene-expression in MDA-MB-231 cells treated with compound 968 were accompanied by the activation of apoptosis, and decreased invasiveness and resistance of cancer cells to chemotherapeutic drug doxorubicin.
Taking into consideration our previous findings that treatment of MDA-MB-231 breast cancer cells with compound 968 caused substantial alterations in metabolically sensitive histone H4K16ac and H3K4me3 marksCitation9 and the evidence that histone modifications are important determinants of gene expression changes,Citation36-Citation38 we investigated if downregulation of cancer-related genes in the MDA-MB-231 cells treated with compound 968 is associated with the gene-specific histone modification changes. The results of our study demonstrate that the majority of downregulated genes were characterized by a decrease in gene-specific histone H3K4me3, a main epigenetic modification linked to activation of transcription.Citation39,Citation40 Therefore, one of the mechanisms of decreased gene expression caused by the compound 968 treatment appears to be the loss of histone H3K4me3 at the corresponding gene promoters. In addition, we show that the loss of histone H3K4me3 at global and gene-specific levels is associated with inhibition of the expression of SETD1 and ASH2L genes. It is well-established that ASH2 is overexpressed in most human tumors and inhibition of ASH2 expression by siRNA markedly decreases cancer cell proliferation.Citation41 Also, our finding of reduced expression of DNMT1, DNMT3B, SETD1A, ASH2L and SIRT in compound 968-treated MDA-MB-231 cells concurs with previous reports demonstrating that the expression of chromatin-modifying genes, including DNMT1, DNMT3B,Citation42 SETD1Citation43 and SIRT1,Citation44 is decreased during apoptosis.
In conclusion, the results of this study provide evidence that the cytotoxicity of compound 968 is associated with a marked reduction of the expression of critical cancer genes, especially anti-apoptotic and metastasis-related oncogenes. Our study also indicates that this downregulation is linked to epigenetic mechanisms that affect the gene-specific level of metabolically sensitive histone modification marks, especially loss of histone H3K4 methylation. Importantly, the results of the present study provide experimental proof of interdependence between cancer cell metabolism and epigenetic regulatory mechanisms and suggests that modification of intracelluar metabolism may enhance the efficiency of epigenetic therapy.
Materials and Methods
Cell lines and treatment with compound 968
MCF-10A, MDA-MB-361, T-47D, Hs-578T, and MDA-MB-231 breast cell lines were obtained from the American Type Culture Collection (ATCC) and maintained according to ATCC’s recommendations. The GLS1 inhibitor compound 968 {5-[3-bromo-4-(dimethylamino)phenyl]-2,2-dimethyl-2,3,5,6-tetrahydrobenzo[a]phenanthridin-4(1H)-one} was purchased from SPECS. In experiments with compound 968, media containing 10 μM Compound 968 and supplemented with 1% fetal bovine serum was added to the cells 5 h after seeding. After 2 and 4 d of treatment, cell survival was determined by counting the viable cells using the Trypan Blue exclusion method. The experiments were repeated and each cell line at each time interval was tested in triplicate.
RNA isolations and quantitative reverse-transcription PCR (qRT-PCR) array analysis
After 2 and 4 d of treatment with compound 968, the cells were harvested by treatment with 0.05% trypsin-EDTA (Invitrogen Corporation), washed in phosphate-buffered saline (PBS), and RNA was isolated immediately. Total RNA was extracted from MDA-MB-231 breast cancer cells using the RNeasy Plus Mini kits (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg RNA, using a RTCitation2 First Strand Kit (Qiagen). The gene expression profiles in untreated and Compound 968-treated MDA-MB-231 cells were determined by qRT-PCR using Human Oncogenes and Tumor Suppressor Genes RTCitation2 Profiler™ PCR Arrays (Qiagen) according to the manufacturer’s protocol.
qRT-PCR analysis
Total RNA (2 μg) was reverse transcribed using random primers and high capacity cDNA archive kits (Applied Biosystems). The following gene expression assays (Applied Biosystems) were used for qRT-PCR: DNMT1 (Hs00154749_m1); DNMT3A (Hs01027166_m1), DNMT3B (Hs00171876_m1), SETD1A (Hs00322315_m1), ASH2L (Hs00983120_m1), EHMT2 (Hs00198710_m1) and SIRT1 (Hs01009005_m1). Reactions were performed in a 96-well assay format using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Each plate contained one experimental gene and a housekeeping gene GAPDH (Hs02758991_g1) and all samples were plated in duplicate. The relative amount of each mRNA transcript was measured using the 2−ΔΔCt method.Citation45
Chromatin immunoprecipitation assay
Formaldehyde cross-linking and chromatin immunoprecipitation (ChIP) assays, with primary antibodies against histone H4K16ac (Millipore) and H3K4me3 (Millipore), were performed by using a Magna Chromatin Immunoprecipitation Assay Kit (Millipore). Purified DNA from immunoprecipitates and from input DNA was analyzed by quantitative PCR (qPCR) with Human Oncogenes and Tumor Suppressor Genes EpiTect ChIP PCR Arrays (Qiagen) according to the manufacturer’s protocol.
Apoptosis assay
Twenty-four hours after treatment of MDA-MB-231 cells with Compound 969, apoptosis was determined by caspase 3/7 activation using Caspase-Glo® 3/7 Assay kit (Promega) according to the manufacturer’s protocol. The protein concentration was determined by Bradford assay (Pierce), and caspase activities for all samples were normalized to an equal protein amount. The experiments were repeated twice, and each cell line tested in triplicate.
Cell invasion assay
Cell invasion activity of MDA-MB-231 cells treated with Compound 968 was assessed by using a BD BioCoat™ Matrigel™ Invasion Chamber (BD Biosciences) according to the manufacturer’s instructions. Cells (5 × 104 cells/ml) were loaded into chamber inserts containing an 8-μm pore size membrane with a thin matrigel matrix layer. Cells migrating to the lower surface of the membrane during 48 h were fixed with 100% methanol. The membranes with migrated cells were then stained with hematoxylin, scanned, and digital images were obtained with an Aperio Scanscope System (Aperio Technologies).
Drug sensitivity assay
To assay drug sensitivity, MDA-MB-231 cells were treated either with DMSO and 0.1, 1.0, or 10.0 μM DOX, or compound 968 and 0.1, 1.0 or 10.0 μM DOX. After 48 h of incubation, the IC50 (inhibitory concentration to produce 50% cell death) values were determined. The experiments were repeated twice, and each cell line tested in triplicate.
Statistical analyses
Results are presented as mean ± SD. Statistical analyses were conducted by one-way analysis of variance, with pair-wise comparisons being conducted by Student-Newman-Keuls test.
| Abbreviations: | ||
| GLS1 | = | glutaminase |
| HDAC9 | = | histone deacetylase 9 |
| HAT | = | histone acetyltransferases |
| DNMT | = | DNA methyltransferase |
| HMT | = | histone methyltransferase |
| HDM | = | histone demethylase |
| H4K16ac | = | H4 lysine 16 acetylation |
| H3K4me3 | = | H3 lysine 4 trimethylation |
| qRT-PCR | = | quantitative reverse-transcription PCR |
| ChIP | = | chromatin immunoprecipitation |
| PBS | = | phosphate-buffered saline |
Additional material
Download Zip (104.7 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/22713
References
- Hitchler MJ, Domann FE. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic Biol Med 2009; 47:115 - 27; http://dx.doi.org/10.1016/j.freeradbiomed.2009.04.010; PMID: 19362589
- Teperino R, Schoonjans K, Auwerx J. Histone methyl transferases and demethylases; can they link metabolism and transcription?. Cell Metab 2010; 12:321 - 7; http://dx.doi.org/10.1016/j.cmet.2010.09.004; PMID: 20889125
- Burgio G, Onorati MC, Corona DF. Chromatin remodeling regulation by small molecules and metabolites. Biochim Biophys Acta 2010; 1799:671 - 80; http://dx.doi.org/10.1016/j.bbagrm.2010.05.007; PMID: 20493981
- Rajendran P, Williams DE, Ho E, Dashwood RH. Metabolism as a key to histone deacetylase inhibition. Crit Rev Biochem Mol Biol 2011; 46:181 - 99; http://dx.doi.org/10.3109/10409238.2011.557713; PMID: 21599534
- Thangaraju M, Gopal E, Martin PM, Ananth S, Smith SB, Prasad PD, et al. SLC5A8 triggers tumor cell apoptosis through pyruvate-dependent inhibition of histone deacetylases. Cancer Res 2006; 66:11560 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-06-1950; PMID: 17178845
- Ferreira LMR, Hebrant A, Dumont JE. Metabolic reprogramming of the tumor. Oncogene 2012; 31:3999 - 4011; http://dx.doi.org/10.1038/onc.2011.576; PMID: 22231450
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683 - 92; http://dx.doi.org/10.1016/j.cell.2007.01.029; PMID: 17320506
- Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 2012; 22:50 - 5; http://dx.doi.org/10.1016/j.gde.2012.02.008; PMID: 22402447
- Simpson NE, Tryndyak VP, Beland FA, Pogribny IP. An in vitro investigation of metabolically sensitive biomarkers in breast cancer progression. Breast Cancer Res Treat 2012; 133:959 - 68; http://dx.doi.org/10.1007/s10549-011-1871-x; PMID: 22101407
- DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313 - 24; http://dx.doi.org/10.1038/onc.2009.358; PMID: 19881548
- Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism?. Oncotarget 2010; 1:734 - 40; PMID: 21234284
- Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol 2012; 23:362 - 9; http://dx.doi.org/10.1016/j.semcdb.2012.02.002; PMID: 22349059
- Wang J-B, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18:207 - 19; http://dx.doi.org/10.1016/j.ccr.2010.08.009; PMID: 20832749
- Asiago VM, Alvarado LZ, Shanaiah N, Gowda GA, Owusu-Sarfo K, Ballas RA, et al. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res 2010; 70:8309 - 18; http://dx.doi.org/10.1158/0008-5472.CAN-10-1319; PMID: 20959483
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009; 324:1076 - 80; http://dx.doi.org/10.1126/science.1164097; PMID: 19461003
- Katt WP, Ramachandran S, Erickson JW, Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther 2012; 11:1269 - 78; http://dx.doi.org/10.1158/1535-7163.MCT-11-0942; PMID: 22496480
- Steward MM, Lee J-S, O’Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol 2006; 13:852 - 4; http://dx.doi.org/10.1038/nsmb1131; PMID: 16892064
- Tate CM, Lee JH, Skalnik DG. CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. FEBS J 2010; 277:210 - 23; http://dx.doi.org/10.1111/j.1742-4658.2009.07475.x; PMID: 19951360
- Robin P, Fritsch L, Philipot O, Svinarchuk F, Ait-Si-Ali S. Post-translational modifications of histones H3 and H4 associated with the histone methyltransferases Suv39h1 and G9a. Genome Biol 2007; 8:R270; http://dx.doi.org/10.1186/gb-2007-8-12-r270; PMID: 18096052
- Vaquero A, Sternglanz R, Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007; 26:5505 - 20; http://dx.doi.org/10.1038/sj.onc.1210617; PMID: 17694090
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 2010; 70:8981 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-10-1666; PMID: 21045145
- Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010; 330:1340 - 4; http://dx.doi.org/10.1126/science.1193494; PMID: 21127244
- Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, et al. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res 2001; 61:4892 - 900; PMID: 11406568
- Zhang M, Guo R, Zhai Y, Yang D. LIGHT sensitizes IFNgamma-mediated apoptosis of MDA-MB-231 breast cancer cells leading to down-regulation of anti-apoptosis Bcl-2 family members. Cancer Lett 2003; 195:201 - 10; http://dx.doi.org/10.1016/S0304-3835(03)00148-4; PMID: 12767529
- Carbone GM, Napoli S, Valentini A, Cavalli F, Watson DK, Catapano CV. Triplex DNA-mediated downregulation of Ets2 expression results in growth inhibition and apoptosis in human prostate cancer cells. Nucleic Acids Res 2004; 32:4358 - 67; http://dx.doi.org/10.1093/nar/gkh744; PMID: 15314206
- Dubská L, Andera L, Sheard MA. HER2 signaling downregulation by trastuzumab and suppression of the PI3K/Akt pathway: an unexpected effect on TRAIL-induced apoptosis. FEBS Lett 2005; 579:4149 - 58; http://dx.doi.org/10.1016/j.febslet.2005.06.047; PMID: 16023111
- Toker A, Yoeli-Lerner M. Akt signaling and cancer: surviving but not moving on. Cancer Res 2006; 66:3963 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-06-0743; PMID: 16618711
- Park EJ, Min HY, Chung HJ, Hong JY, Kang YJ, Hung TM, et al. Down-regulation of c-Src/EGFR-mediated signaling activation is involved in the honokiol-induced cell cycle arrest and apoptosis in MDA-MB-231 human breast cancer cells. Cancer Lett 2009; 277:133 - 40; http://dx.doi.org/10.1016/j.canlet.2008.11.029; PMID: 19135778
- Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011; 11:558 - 72; http://dx.doi.org/10.1038/nrc3090; PMID: 21734724
- Lin Y, Cui H, Xu H, Yue L, Xu H, Jiang L, et al. Jolkinolide B induces apoptosis in MDA-MB-231 cells through inhibition of the PI3K/Akt signaling pathway. Oncol Rep 2012; 27:1976 - 80; PMID: 22426554
- Haas-Kogan DA, Kogan SC, Levi D, Dazin P, T’Ang A, Fung YK, et al. Inhibition of apoptosis by the retinoblastoma gene product. EMBO J 1995; 14:461 - 72; PMID: 7859736
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006; 444:61 - 6; http://dx.doi.org/10.1038/nature05194; PMID: 17080083
- Tryndyak VP, Beland FA, Pogribny IP. E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int J Cancer 2010; 126:2575 - 83; PMID: 19839049
- Zhang Y, Pu X, Shi M, Chen L, Qian L, Song Y, et al. c-Jun, a crucial molecule in metastasis of breast cancer and potential target for biotherapy. Oncol Rep 2007; 18:1207 - 12; PMID: 17914574
- Engelmann D, Pützer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res 2012; 72:571 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-11-2575; PMID: 22298593
- Kwon MJ, Kim SS, Choi YL, Jung HS, Balch C, Kim SH, et al. Derepression of CLDN3 and CLDN4 during ovarian tumorigenesis is associated with loss of repressive histone modifications. Carcinogenesis 2010; 31:974 - 83; http://dx.doi.org/10.1093/carcin/bgp336; PMID: 20053926
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13:343 - 57; http://dx.doi.org/10.1038/nrg3173; PMID: 22473383
- Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693 - 705; http://dx.doi.org/10.1016/j.cell.2007.02.005; PMID: 17320507
- Sims RJ 3rd, Reinberg D. Histone H3 Lys 4 methylation: caught in a bind?. Genes Dev 2006; 20:2779 - 86; http://dx.doi.org/10.1101/gad.1468206; PMID: 17043307
- Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 2012; 13:297 - 311; PMID: 22473470
- Lüscher-Firzlaff J, Gawlista I, Vervoorts J, Kapelle K, Braunschweig T, Walsemann G, et al. The human trithorax protein hASH2 functions as an oncoprotein. Cancer Res 2008; 68:749 - 58; http://dx.doi.org/10.1158/0008-5472.CAN-07-3158; PMID: 18245475
- Kassis ES, Zhao M, Hong JA, Chen GA, Nguyen DM, Schrump DS. Depletion of DNA methyltransferase 1 and/or DNA methyltransferase 3b mediates growth arrest and apoptosis in lung and esophageal cancer and malignant pleural mesothelioma cells. J Thorac Cardiovasc Surg 2006; 131:298 - 306; http://dx.doi.org/10.1016/j.jtcvs.2005.05.022; PMID: 16434257
- Lee J-H, You J, Dobrota E, Skalnik DG. Identification and characterization of a novel human PP1 phosphatase complex. J Biol Chem 2010; 285:24466 - 76; http://dx.doi.org/10.1074/jbc.M110.109801; PMID: 20516061
- Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Lüscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A 2012; 109:E187 - 96; http://dx.doi.org/10.1073/pnas.1105304109; PMID: 22190494
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101 - 8; http://dx.doi.org/10.1038/nprot.2008.73; PMID: 18546601