Abstract
Hepatocellular carcinoma (HCC) incidence has increased in the US and also has one of the fastest growing death rates of any cancer. The purpose of the current study was to discover novel genome-wide aberrant DNA methylation patterns in HCC tumors that are predominantly HCV-related. Infinium HumanMethylation 450K BeadChip arrays were used to examine genome-wide DNA methylation profiles in 66 pairs of HCC tumor and adjacent non-tumor tissues. After Bonferroni adjustment, a total of 130,512 CpG sites significantly differed in methylation level in tumor compared with non-tumor tissues, with 28,017 CpG sites hypermethylated and 102,495 hypomethylated in tumor tissues. Absolute tumor/non-tumor methylation differences ≥ 20% were found in 24.9% of the hypermethylated and 43.1% of the hypomethylated CpG sites; almost 10,000 CpG sites have ≥ 30% DNA methylation differences. Most (60.1%) significantly hypermethylated CpG sites are located in CpG islands, with 21.6% in CpG shores and 3.6% in shelves. In contrast, only a small proportion (8.2%) of significantly hypomethylated CpG sites are situated in islands, while most are found in open sea (60.2%), shore (17.3%) or shelf (14.3%) regions. A total of 2,568 significant CpG sites (2,441 hypermethylated and 127 hypomethylated) covering 589 genes are located within 684 differentially methylated regions defined as regions with at least two significant CpG sites displaying > 20% methylation differences in the same direction within 250-bp. The top 500 significant CpG sites can significantly distinguish HCC tumor from adjacent tissues with one misclassification. Within adjacent non-tumor tissues, we also identified 75 CpG sites significantly associated with gender, 228 with HCV infection, 17,207 with cirrhosis, and 56 with both HCV infection and cirrhosis after multiple comparisons adjustment. Aberrant DNA methylation profiles across the genome were identified in tumor tissues from US HCC cases that are predominantly related to HCV infection. These results demonstrate the significance of aberrant DNA methylation in HCC tumorigenesis.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the third leading cause of cancer-associated deaths in men worldwide.Citation1 An increase in HCC incidence has been observed in the US recently, where HCC has one of the fastest growing death rates for any cancer in both men and women.Citation2,Citation3 The initiation and development of HCC is a multi-step process with accumulated genetic and epigenetic alterations resulting from diverse etiologies, including hepatitis B (HBV) and hepatitis C virus (HCV) infections, alcohol abuse, non-alcoholic steatohepatitis, and exposure to environmental carcinogens, such as aflatoxin B1 (AFB1). These aberrant biological changes may lead to the activation of oncogenes and inactivation of tumor suppressive genes, which are among the key regulators of hepatocarcinogenesis.
Altered DNA methylation (hyper- or hypo-methylation) is one of the early events and the most consistent epigenetic change observed in HCC. Global DNA hypomethylation, particularly in repeated sequences and transposable elements, largely affects intergenic and intronic regions of the genome and plays a critical role in increasing chromosomal instability.Citation4 Hypermethylation of promoter regions of crucial genes (such as tumor suppressor, DNA repair and cell cycle control genes) can repress relevant gene expression and downregulate biological functions and is associated with an increased risk of HCC.Citation5 Aberrant DNA methylation has been frequently found in CpG islands, as well as CpG shores (regions up to 2 kb from the islands) and CpG shelves (2–4 kb from the islands),Citation6 and may contribute to tumorigenesis.Citation7-Citation9
Prior studies have used a range of methods to investigate methylation in 1,500 to 27,000 CpG sites in HCC tumor/adjacent tissues from Japan,Citation10,Citation11 Korea,Citation12 France,Citation13 GermanyCitation14 and the USCitation15 but with relatively small sample sizes of 5 to 38 cases. Our own prior study used the Illumina HumanMethylation 27K arrays to investigate methylation in 62 Taiwan HCC cases, predominantly with HBV infection (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37988).Citation16 While a powerful tool, the 27K array only provides information on 27,578 CpG sites representing a small part of the entire genome and mainly covers promoter regions.Citation14,Citation17 Four recent reports demonstrate the accuracy and reproducibility of the Infinium HumanMethylation 450K BeadChip arrays that more comprehensively examines genome-wide DNA methylation profiles. It includes 485,764 CpG sites from 21,231 genes covering 96% of CpG islands and 99% of RefSeq genes with multiple probes per gene from the UCSC database.Citation6,Citation18-Citation20 The overall congruence with pyrosequencing data are also very good.Citation20 The 450K BeadChips span not only CpG islands, but also CpG shores and CpG shelves. Application of the arrays to analyze DNA from cancer cell lines and tissues indicate that it is an efficient, robust and affordable tool in assessing epigenetic changes across the human genome.Citation6,Citation18-Citation20 In the current study, we applied these arrays to determine aberrant DNA methylation in predominantly HCV-related HCC in the US. The identification of novel DNA methylation markers contributes to our understanding of HCC pathogenesis, and should lead to improvements in early diagnosis and treatment.
Results
Clinical and pathological characteristics of HCC patients
Clinical and pathological characteristics are described in . The average age at HCC diagnosis is 59.5 ± 14.6 y. Most patients are male (76%) and Caucasian (52%). Approximately 20% of cases are positive for HBV and 29% are positive for HCV. Twenty-nine percent are negative for both HBV and HCV and 6% are positive for both HBV and HCV. A total of 36 cases (55%) smoked and 36 cases (55%) consumed alcohol. Among the 66 HCC patients, 73% have pathologically defined cirrhosis and 61% have tumors grade III or IV.
Table 1. Clinical and pathological characteristics of 66 US HCC patients
Comparison of β–value density by the two chemical assays in Infinium Methylation 450K technology
The design of Infinium Methylation 450K arrays comprises two chemical assays (Infinium I and Infinium II), which may differ in their sensitivity for detecting extreme methylation values (i.e., 0 and 1) as previously reported.Citation19 We compared the distribution of β-value density between the two chemical assays. The β-value density plots display a bimodal distribution for Infinium I and Infinium II assays (Fig. S1A). Consistent with previous observations, there are two peaks for less methylated and extensively methylated CpG sites.Citation19 A shift toward the center of methylation was observed for the Infinium II assay data (Fig. S1C) compared with that of the Infinium I assay (Fig. S1B), and the same shifting pattern was observed for tumor and adjacent non-tumor tissues. Because we conducted probe-level analyses of the differences between tumor tissues and adjacent normal tissues, any shift between the two assays will not impact differences.
Genome-wide DNA methylation profiles differentiate HCC tumor from non-tumor tissues
Methylation of 130,512 (27.5%) CpG sites out of 473,929 across the entire genome (covering 13,790 genes) significantly differed between HCC tumor and adjacent non-tumor tissues after Bonferroni correction for multiple comparisons. Among all significant CpG sites, 28,017 (21%) are significantly hypermethylated (covering 5,774 genes), and 102,495 (79%) are significantly hypomethylated (covering 11,606 genes) in HCC tumor tissues. A volcano plot displaying the mean DNA methylation differences for all CpG sites is given in Fig. S2. These data confirm that aberrant DNA methylation is a very common event in HCC tumor tissues across the entire genome with hypomethylation of CpG sites being more frequently observed than hypermethylation.
The frequencies of all significant CpG sites by tumor/non-tumor differences are shown in Table S1. Twenty five percent of the hypermethylated CpG sites (6,969) and 43.1% of the hypomethylated CpG sites (44,162) have an absolute tumor/non-tumor methylation difference ≥ 20%. Almost 10,000 CpG sites (< 10% of all significant sites) have a ≥ 30% difference in DNA methylation between tumor and non-tumor tissues. These data suggest that DNA methylation changes in HCC tumor tissues are robust and may be excellent candidate biomarkers. The genomic regions of the significantly hyper- or hypomethylated CpG sites are distributed differently (Table S2). Sixty percent (16,841) of the CpG sites significantly hypermethylated are in CpG island regions, with 21.6% in CpG shores and 3.6% in shelf regions. In contrast, only a small proportion (8.2%) of the significantly hypomethylated CpG sites (8,412) are situated in islands, while most are found in open sea (60.2%), shore (17.3%) or shelf (14.3%) regions, suggesting a potential role in genomic instability.
After Bonferroni adjustment, there are many more significant CpG sites identified in male HCC (87,305 hyper- and 19,530 hypomethylation CpG sites) compared with female (1,492 hyper- and 356 hypomethylation CpG sites) possibly due to the larger number of male (50) then female (16) cases. Most significant CpG sites found in female HCC (1,489 hypermethylated and 339 hypomethylated) overlap with those in male (Fig. S3). For male HCC, there are, respectively, 1,888 and 103 significant CpG sites located on the X and Y chromosomes. Only 4 hypermethylated CpG sites on the X chromosome are significantly different between female HCC tumor and non-tumor tissues (data not shown).
Biological characteristics of the top significant differentially methylated CpG sites and differentially methylated regions (DMRs)
Using more stringent criteria (see Methods) to reduce the potential impact of extreme β value on methylation difference and to identify potentially biologically important CpG sites (≥ 20% methylation difference), we identified 3,921 significantly hypermethylated CpG sites (covering 1,112 genes), and 696 significantly hypomethylated CpG sites (covering 169 genes) for further analysis (Table S3). shows a hierarchical cluster analysis of the top 500 significant CpG sites (based on statistical significance) that distinguish HCC tumor from adjacent tissues. Excellent separation of tumor and adjacent tissues is observed with only one misclassification. shows that > 82% of the hypermethylated CpG sites (3,220) are in CpG islands, with fewer in CpG shore (554), shelf (22) and open sea (125) regions. Significantly hypomethylated CpG sites are more common in open sea (66.7%) than island (9.9%), shore (13.8%) or shelf (9.6%) regions.
Figure 1. Hierarchical cluster analysis of the top 500 significantly differentially methylated CpG sites between 66 pairs of tumor and adjacent tissues. Blue represents tumor tissue and red represents adjacent non-tumor tissue. The top 500 significant CpG sites can clearly distinguish HCC tumor from adjacent tissues with only one misclassification.
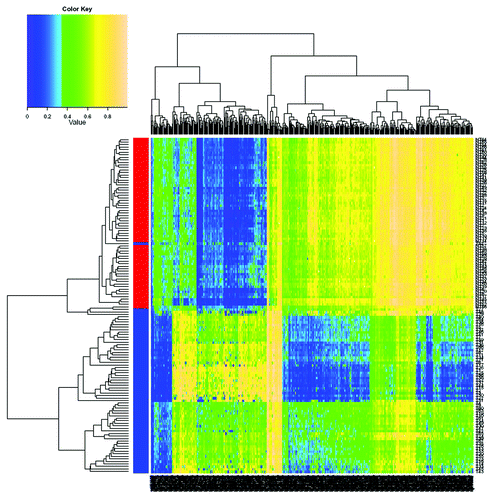
Table 2. Distribution of genomic regions for significant CpG sites passing the stringent filtering criteria
There are 2,109 (53.8%) significantly hypermethylated CpG sites located within promoter regions and 411 (10.5%) in regions with high CpG content (Table S4). There are relatively few hypomethylated CpG sites located within promoters (19.5%), and none in regions with high CpG content. More than 62% (2,441/3,921) of significantly hypermethylated CpG sites are located within 839 DMRs defined as regions with at least two significant CpG sites displaying > 20% methylation differences in the same direction within 250 bp.Citation21 A total of 569 genes (with 2 to 29 CpG sites per gene) harbor 655 hypermethylated DMRs. Others (184 DMRs) are located within non-coding regions. There are 127 (18.3%) significantly hypomethylated CpG sites within 56 DMRs, which include 29 DMRs covering 20 genes (with 2 to 16 CpG sites per gene), and 27 DMRs located within non-coding regions. The genome-wide distribution of significant CpG sites within DMRs is shown in Figure S4. The hypermethylated DMRs (blue lines) are widely distributed across the whole genome with many obvious clusters on different chromosomes, while the hypomethylated DMRs (red lines) are located within a limited number of chromosomes with a few significant clusters observed in chromosomes 5, 7 and 8. These data suggest that significantly hypermethylated DMRs are more common in HCC tumor tissues compared with hypomethylated DMRs, which is the reverse of the observation for single CpG sites that are more frequently hypomethylated in HCC tumor tissue (Fig. S2). The top 20 significantly hyper- or hypomethylated CpG sites within DMRs are listed in ranked by statistical significance.
Table 3. Top 20 significant CpG sites and genes within DMRs in HCC tumor tissues compared with adjacent non-tumor tissues
DNA methylation patterns affected by HCC risk factors
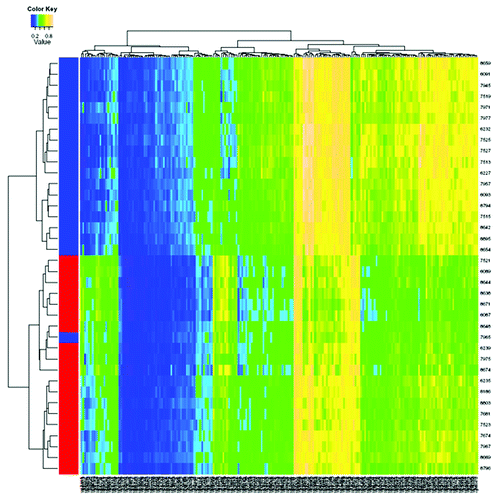
Two-sample t-test was used to compare methylation differences by HCC risk factors within adjacent non-tumor tissues. After Bonferroni correction, 75 CpG sites are significantly associated with gender, but unrelated to other risk factors. No sites significantly differ by alcohol use or HBV infection status. Methylation level at one CpG in OAZ3 (MRPL9) significantly differs between ever and never smokers. A total of 228 CpG sites are significantly associated with HCV infection, and 17,207 CpG sites are significantly correlated with cirrhotic status, with 5,398 hypermethylated and 11,809 hypomethylated in cirrhotic tissues. shows unsupervised hierarchical clustering analysis for 228 HCV-related CpG sites, and Figure S5 shows the top 500 differentially methylated CpG sites between cirrhotic and non-cirrhotic tissues. These significant aberrant CpG methylation sites are generally able to well separate HCV positive from negative tissues and cirrhotic from non-cirrhotic tissues, with some misclassifications (1–12 tissues). We next compared the specificity of the significant CpG sites related to HCC and different risk factors by Venn diagram and found no overlapping CpG sites between tumor and risk factors of gender (male) and HCV infection. Only 8 CpG sites are significantly associated with both HCC tumor and cirrhotic tissues (Fig. S6A), indicating the potentially independent roles of aberrant DNA methylation in hepatocarcinogenesis. A few (56) CpG sites are significantly correlated with both HCV infection and cirrhosis.
Figure 2. Hierarchical cluster analysis of the 228 methylated CpG sites that significantly differentiate HCV positive and HCV negative HCC in adjacent non-tumor tissues among cases who are HBV negative. Blue represents HCV positive, and red represents HCV negative. The significant aberrant CpG sites are able to successfully separate HCV positive from negative tissues with only one misclassification.
Comparisons of significant CpG sites identified by Infinium Methylation 27K and 450K data
Comparing the significant hyper- and hypo-methylated CpG sites previously identified using the Infinium Methylation 27K arrays in 62 Taiwanese HCCCitation16 and the 450K arrays in 66 US HCC patients (Fig. S6B), we found an overlap of 146 significant hypermethylated CpG sites (covering 128 genes) and 15 hypomethylated CpG sites (covering 15 genes). These are promising candidates for further confirmation in future studies. All overlapping CpG sites are located within specific genes, with 113 CpG sites (70.2%) located in island, 24 (14.9%) in shore, 1 (0.6%) in shelf, and 23 (14.3%) in open sea regions. However, no genes have both aberrant hyper- and hypomethylation suggesting potentially independent biological mechanisms involved in those two types of epigenetic changes.
Discussion
We use lllumina HumanMethylation 450K arrays to identify genome-wide aberrant DNA methylation profiles that contribute to predominantly HCV-related HCC in the US. A total of 130,512 CpG sites across the entire genome significantly differ in methylation levels between HCC tumor and adjacent non-tumor tissues after Bonferroni correction for multiple comparisons. More CpG sites (102,495, 79%) in HCC tumor tissues are hypomethylated than hypermethylated (28,017, 21%), which is consistent with the proportion observed in previous HCC genome-wide methylation studies,Citation10-Citation14 including ours.Citation16 However, these previous studies lacked data on DMRs. Our current study suggests that, in contrast to specific CpG sites, significant hypermethylated DMRs are more common in HCC tumor tissue compared with hypomethylated DMRs. Using DMRs rather than CpG sites may be more accurate and biologically relevant, and minimize potential artifacts due to random methylation alterations in single CpG sites.
The use of cost-effective, high-throughput techniques allows a hypothesis-free approach to discover novel DNA methylation markers in HCC. An earlier study examined 6,458 CpG islands in 38 paired HCC tumor and adjacent non-tumor tissues from Japan using methylated CpG island amplification microarrays.Citation10 A total of 719 (11%) genes were identified as hypermethylated in HCC. Another study was performed to screen methylated genes in HCC tissues using methylated CpG island amplification coupled with CpG island microarray analysis (containing 15,134 probes covering 6,157 unique genes). The authors identified 332, 342, and 259 genes that were, respectively, aberrantly methylated in HBV-positive, HCV-positive and HBV/HCV-negative HCC tissues. Four of these genes (KLHL35, PAX5, PENK and SPDYA) were significantly hypermethylated in HCC tissue irrespective of viral status;Citation11 current results confirm this observation. Two studies used Illumina 1,500 Golden Gate arrays to screen DNA methylation patterns on 5 paired HCC tissues from KoreaCitation12 and 30 from France.Citation13 The first study identified 24 new genes as potentially novel methylation markers for HCC, and confirmed four hypermethylated genes within tumor tissues by the inactivation of gene expressions.Citation12 In the second study, 124 CpG sites in 94 genes were differentially methylated between tumors and surrounding tissues with 34 hypermethylated and 90 hypomethylated.Citation13 Ammerpohl et al. investigated genome-wide DNA methylation profiles using Illumina HumanMethylation 27K array in 12 HCC, 15 cirrhotic and 12 normal tissues. They found that 998 GpG sites were differentially hypomethylated while 278 sites were hypermethylated in HCC as compared with normal tissues. In the same study, comparing HCC and cirrhotic tissues, 1,050 CpG sites were significantly hypomethylated and 378 sites were hypermethylated in the tumor tissue, while only 8 CpG sites (covering 8 genes SPRR3, TNFSF15, ALOX12, ANGPTL7, CELSR1, CRMP1, GNRH2 and LOC55908) were differentially methylated between both HCC vs. cirrhosis and cirrhosis vs. normal tissues comparisons.Citation14 We previously utilized Illumina HumanMethylation 27K arrays with the largest sample size prior to the work presented here (62 pairs of Taiwanese HCC tumor and adjacent non-tumor tissues) and identified many more CpG sites that significantly differentiate tumor tissues from adjacent tissues than existing studies.Citation17 In our prior study, a total of 2,324 CpG sites significantly differed (with 684 CpG sites significantly hypermethylated and 1,640 hypomethylated) in HCC tumor compared with non-tumor tissues,Citation17 including most previously identified novel genes or CpG sites.Citation12-Citation14
We also observe high percentages of CpG sites (25% hypermethylated and 43% hypomethylated) with large tumor/non-tumor methylation differences (≥ 20%). Our ultimate goal is to identify a panel of aberrant DNA methylation markers that can be detected in easily accessible biological materials, such as blood, to improve early diagnosis of HCC. Therefore, to select a small number of promising, biologically important CpG sites, DMRs or genes, we applied more stringent criteria to reduce the potential impact of extreme β values on methylation differences. Ultimately, 3,921 (84.9%) significantly hypermethylated and 696 (15.1%) hypomethylated CpG sites were identified (Table S3). A total of 2,441 significantly hypermethylated and 127 hypomethylated CpG sites are located within DMRs covering 530 genes with 2 to 29 significant CpG sites per gene.
We believe that the use of stringent criteria to select promising CpG sites/DMRs/genes, together with the large sample size, are unique strengths of the current study, which allowed us to identify novel aberrant DNA methylation profiles in critical genes potentially applicable to improving early diagnosis of HCC. Importantly, there is an overlap of 161 significant CpG sites covering 143 genes in the two independent HCC studies using Infinium Methylation 27K and 450K arrays, indicating a crucial role for those genes in hepatocarcinogenesis. These data further confirm that not only is aberrant DNA methylation across the entire genome a common event in HCC tumor tissues, but also large tumor/non-tumor methylation and DMRs differences are usually observed. Using these aberrant DNA methylation markers, we were able to successfully classify HCC tumor and non-tumor tissues with only one misclassification (), suggesting their promise for future clinical application. However, Robinson et al.Citation21,Citation22 found that genetic variation, including single nucleotide polymorphisms (SNP) and copy number variants (CNV) associated with HCC, significantly confounded DNA methylation differences obtained from genome-wide analyses. They suggested the exclusion of CpG sites that overlap with SNPs or are located within CNVs regions according to the existing genomic databases to reduce potential artifacts. Therefore, simultaneously examining genetic variants and other epigenetic changes that may potentially impact DNA methylation alterations is necessary to elucidate their role in influencing DNA methylation and is the next step of our research. Of course, further functional validation to distinguish real “drivers” of malignant transformation is warranted.
The genomic distribution of significant CpG sites reveals that significantly hypermethylated CpG sites are more frequently located at CpG islands (60.1%) and shores (21.6%), while most significant hypomethylated CpG sites (60.2%) are located in open sea regions (Table S2). These data are consistent with the findings from a previous colorectal cancer cells studyCitation6 and our Taiwan HCC study.Citation16 It is well known that epigenetic inactivation of genes in malignant tumors including HCC is largely based on transcriptional silencing by aberrant CpG island methylation in promoter regions and that genomic instability is believed to be related to global hypomethylation. However, the location of the analyzed CpG sites may also be important in HCC specificity. For example, Jain et al. found that the 5′ region up to 48 nt from the transcription start site of GSTP1 is selectively methylated in HCC, whereas the 3′ region is methylated in all liver tissues, including normal liver.Citation23 From a functional genome standpoint, the different distributions of significant hypermethylated and hypomethylated CpG sites suggest important epigenetic mechanism in hepatocarcinogenesis, and provide extensive information on the relationship between the development of human cancer and the DNA methylation landscape.
Differences in methylation patterns by HCC risk factors were also explored in adjacent non-tumor tissues. We found that a few CpG sites are significantly associated with gender, age, cigarette smoking, alcohol drinking or HBV infection. Until now, methylation in only a few genes (CDH1, Gadd45beta, p16, RASSF1 and STAT1) have been reported to be correlated with HCV infection by a candidate gene approach.Citation24-Citation29 Using a genome-wide approach, for the first time, we identified a total of 228 aberrantly methylated CpG sites (covering 147 genes) that are significantly associated with HCV infection in adjacent non-tumor liver tissue. We did not observe any overlapping CpG sites related to both HCC and HCV, suggesting that the aberrantly methylated CpG sites associated with HCV are not necessarily related to hepatocarcinogenesis. This is consistent with a previous genome-wide study that found a number of genes (169 probes) commonly methylated in HCC irrespective of hepatitis virus status,Citation11 suggesting potentially independent patterns of aberrant DNA methylation for HCV infection and malignant transformation. We ascertained that 17,207 CpG sites are significantly correlated with cirrhotic liver tissues across the genome, including 56 CpG sites that overlapped with those associated with HCV and 8 CpG sites that overlapped with those associated with HCC (Fig. S6A). One previous genome-wide study identified 47 differentially methylated CpG loci between cirrhotic and non-cirrhotic liver tissues; 9 genes overlap with our findings (ACSS1, APC, BACH1, CCDC37, ESR1, GFRA3, KCNQ4, PODN and RASSF5).Citation14 Principal component analysis (PCA) shows that the cirrhotic samples, in general, are on the same major branch of the tree as the non-cirrhotic adjacent tissues and consequently are more similar to the adjacent tissues than to the tumors.Citation14 Another study identified 21 CpG sites significantly differentially methylated between non-tumor cirrhotic tissue of HCC patients and cirrhotic tissues of patients without HCC using Illumina GoldenGate Methylation BeadArray.Citation15 These results support our observations that only a few CpG sites overlap between HCC tumor and cirrhotic tissues, while more overlap is observed between HCV infection and cirrhotic tissues. Previous bioinformatics and biochemical analyses revealed that the enhancer of secreted phosphoprotein 1 (Spp1), a gene associated with fibrosis, is hypomethylated and its expression is upregulated.Citation30 Our data also indicates that two CpG sites of Spp1 are significantly hypomethylated in cirrhotic liver tissues, suggesting DNA hypomethylation of Spp1 may be involved in liver cirrhosis. Three studies also observed overexpressed Spp1 in HCC compared with normal liver tissues, suggesting a crucial role of Spp1 in HCC development.Citation31-Citation33 But we did not observe methylation of any CpG sites in Spp1 associated with HCC tumor tissue. Four different patterns have been proposed by Kanai to describe genome-wide DNA methylation alterations occurring during multistage hepatocarcinogenesis developing from chronic hepatitis to cirrhosis, dysplastic nodule and finally, HCC.Citation34 They include DNA methylation (1) altered in chronic hepatitis/liver cirrhosis, but not in HCC; (2) occur in chronic hepatitis/liver cirrhosis, and are further altered in HCCs; (3) altered in chronic hepatitis/liver cirrhosis, but return to normal in HCCs and (4) altered only in HCCs. Our observations of HCV or cirrhosis-related methylation changes provide support for this theory. However, our results are obtained after diagnosis and thus cannot unravel the potential role of the tumor itself on changing methylation levels. Therefore, prospective analysis of DNA methylation alterations in hepatitis or cirrhotic tissues prior to HCC occurrence may provide a more logical and appropriate substrate to assess their biological relationships with those risk factors.Citation35
In summary, we have comprehensively characterized genome-wide DNA methylation patterns occurring in HCC, and identified a large subset of CpG sites/DMRs/genes correlated with HCV infection, liver cirrhosis or HCC. We believe the robust data obtained from the current large study provides valuable information to better understand the molecular mechanisms involved in multistep of HCC. The clinical application of sorafenib (the only FDA approved anti-angiogenic medication) for advanced HCC patients, and the ongoing clinical trials for IGF modulators and PI3 kinase inhibitors provide promising data for insights into the value of aberrant CpG sites/DMRs/genes that are involved in those biological pathways.Citation36 Further functional studies and follow-up evaluations to clarify the real “drivers” of tumorigenesis among the aberrant DNA methylation markers should have significant clinical application in improving HCC early diagnosis and contribute to effective personalized therapies.
Methods
HCC subjects and specimens
This study was approved by the Institutional Review Board of Columbia University Medical Center. Sixty-six frozen HCC tissues were collected by the Center for Liver Disease and Transplantation and stored in the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center. Histological evaluation of hematoxylin and eosin (H.E.) stained 4 micron thick sections of frozen tissue store at -20°C, for liver tumor and adjacent non-tumor tissues, included assessment of presence, viability and percent of tumor. Tumor samples were macrodissected to ensure > 80% purity of tumor. To insure the DNA extracted from adjacent normal tissue did not contain any tumor cells, tissue sections were cut from frozen tissues and H.E. stained. The stained sections were carefully observed under a microscope by the study pathologist (H.R.) to ensure no tumor tissues or cells were present in the whole sections. Then several sections were cut from the same tissues for DNA extraction. Frozen tissue blocks of adjacent tissue were also evaluated with respect to the presence or absence of cirrhosis. Information on viral infection (HBV, HCV) and clinicopathological features including α-fetoprotein levels, tumor size, tumor number, tumor differentiation, vascular invasion, and capsular infiltration are obtained from the medical records. HBV (HBsAg) and HCV (anti-HCV) status were determined by immunoassay.
DNA preparation and Infinium Methylation 450K assay
DNA was extracted from 66 frozen tumor/adjacent tissues by standard proteinase K/RNase treatment and phenol/chloroform extraction. Bisulfite modification of 1µg DNA was conducted using an EZ DNA Methylation Kit (Zymo Research) according to the manufacturer’s procedure. The Infinium Methylation 450K assay was performed according to Illumina’s standard protocol. Six HCC tumor/adjacent non-tumor tissue pairs were processed on the same chip to avoid chip-to-chip variation.
Statistical methods
Infinium Methylation data were processed with the Methylation Module of GenomeStudio software using HumanMethylation450 manifest v1.1. Methylation levels of CpG sites were calculated as β-values (β = intensity of the methylated allele (M)/[intensity of the unmethylated allele (U) + intensity of the methylated allele (M) + 100].Citation6 For quality control (QC), methylation measures with a detection p-value > 0.05 and samples with a CpG coverage < 95% were removed. Ultimately 485,577 CpG sites in all 66 pairs of samples passed the coverage criteria. Because gender is a risk factor for HCC, and the numbers of male and female cases are uneven (50 vs. 16), potential bias may exist related to significant CpG sites on Y and X chromosomes. Therefore, CpG sites (11,648) on the sex chromosomes were removed for the initial analysis to avoid gender-specific methylation bias, which left 473,929 autosomal CpG sites. CpG sites on the sex chromosomes were included for analysis of gender-specific methylation differences.
Paired t-tests with Bonferroni correction for multiple testing were used to identify CpG sites that are differentially methylated between tumor and adjacent non-tumor tissues. A significant difference was defined as sites with a Bonferroni-corrected p value ≤ 0.05 which corresponds to a raw p value of ≤ 1.06 × 10−7. A volcano plot was used to display mean DNA methylation differences for all 473,929 CpG sites. Hierarchical clustering of the methylation data was performed with the top significant sites to cluster tissue status (tumor vs. non-tumor) or HCC risk factors (gender, HBsAg, anti-HCV).
Because > 25% of the total CpG sites are statistically significantly different between tumor and adjacent non-tumor tissues, we applied additional filtering criteria to select candidate CpG sites that have large tumor/non-tumor differences in methylation levels. These further filtered sites are more likely to be helpful in the identification of more biologically meaningful methylation changes, which may lead to improvements in identifying biomarkers for early diagnosis and treatment. For significant hypermethylation sites, we selected CpG sites with the following three criteria: (1) the mean difference in methylation level between tumor and adjacent tissues is > 20%; (2) > 70% of the tumor tissues have methylation levels greater than 2 standard deviations (SDs) above the mean methylation level of all 66 adjacent tissues; and (3) the mean methylation level for adjacent tissues is < 25%.Citation16 For significantly hypomethylated sites, we selected CpG sites with the following two criteria: (1) the mean difference in methylation level between adjacent and tumor tissues is > 20%; and (2) > 70% of the adjacent tissues have methylation levels greater than 2 SDs above the mean methylation level of all 66 tumor tissues. In total, 3,921 significantly hypermethylated and 696 hypomethylated CpG sites were identified.
To check the robustness of the filtered lists of CpG sites, we conducted a 3-fold cross-validation on the hypomethylated CpG sites, where we randomly choose 44 out of 66 pairs to form a training set and the remaining 22 pairs as a testing set. We then repeated the paired t-test using the training set and selected the top 1,000 most significantly hypomethylated CpG sites with the following loosened two criteria to ensure selection of enough candidate CpG sites at each cross-validation: (1) the mean difference in methylation levels between adjacent and tumor tissues is > 20%; (2) > 60% of the adjacent tissues have methylation levels greater than 2 SDs above the mean methylation level of the 44 tumor tissues. We repeated the 3-fold cross-validation 2,000 times and selected the top most frequently selected CpG sites with the same number as in the original list, i.e., 696 hypomethylated CpG sites. The two panels of 696 CpG sites have 622 overlapping sites. Moreover, the 696 top frequently selected CpG sites based on the 2,000 3-fold cross-validations all have selection probability > 60%. Among these CpG sites, we identified differentially methylated regions (DMRs) defined as regions having at least two significant neighboring CpG sites displaying > 20% methylation differences in the same direction within a 250 bp region.Citation22 Genes with at least one DMR are considered as aberrantly methylated genes. The chromosome distribution of significant DMRs was generated by Idiographica web-based software based on human (HG19) build.Citation37
To investigate whether methylation levels are affected by HCC risk factors such as gender (male/female), HBV (positive/negative), HCV (positive/negative), cigarette smoking (ever/never) or alcohol consumption (ever/never), we compared methylation differences by these factors within adjacent non-tumor tissues using a two-sample t-test with Bonferroni correction for multiple testing. Only subjects with complete viral status data were used for investigating associations with viral status.
All analyses were conducted using the R language (www.r-project.org/).
| Abbreviations: | ||
| HCC | = | hepatocellular carcinoma |
| HBV | = | hepatitis B virus |
| HCV | = | hepatitis C virus |
| AFB1 | = | aflatoxin B1 |
| DMRs | = | differentially methylated regions |
Additional material
Download Zip (3.2 MB)Acknowledgments
This work is supported by NIH grants R01 ES005116, P30 ES009089, R03 CA156629, R03 CA150140 and P30 CA013696.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/23062
References
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893 - 917; http://dx.doi.org/10.1002/ijc.25516; PMID: 21351269
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132:2557 - 76; http://dx.doi.org/10.1053/j.gastro.2007.04.061; PMID: 17570226
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10 - 29; http://dx.doi.org/10.3322/caac.20138; PMID: 22237781
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300:455; http://dx.doi.org/10.1126/science.1083557; PMID: 12702868
- Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol 2008; 14:1741 - 8; http://dx.doi.org/10.3748/wjg.14.1741; PMID: 18350605
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011; 98:288 - 95; http://dx.doi.org/10.1016/j.ygeno.2011.07.007; PMID: 21839163
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet 2009; 41:1350 - 3; http://dx.doi.org/10.1038/ng.471; PMID: 19881528
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178 - 86; http://dx.doi.org/10.1038/ng.298; PMID: 19151715
- Ogoshi K, Hashimoto S, Nakatani Y, Qu W, Oshima K, Tokunaga K, et al. Genome-wide profiling of DNA methylation in human cancer cells. Genomics 2011; 98:280 - 7; http://dx.doi.org/10.1016/j.ygeno.2011.07.003; PMID: 21821115
- Gao W, Kondo Y, Shen L, Shimizu Y, Sano T, Yamao K, et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis 2008; 29:1901 - 10; http://dx.doi.org/10.1093/carcin/bgn170; PMID: 18632756
- Shitani M, Sasaki S, Akutsu N, Takagi H, Suzuki H, Nojima M, et al. Genome-wide analysis of DNA methylation identifies novel cancer-related genes in hepatocellular carcinoma. [Epub ahead of print] Tumour Biol 2012; 33:33 - 40; PMID: 21931992
- Shin SH, Kim BH, Jang JJ, Suh KS, Kang GH. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array. J Korean Med Sci 2010; 25:1152 - 9; http://dx.doi.org/10.3346/jkms.2010.25.8.1152; PMID: 20676325
- Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, Gouysse G, McKay-Chopin S, Tavtigian SV, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One 2010; 5:e9749; http://dx.doi.org/10.1371/journal.pone.0009749; PMID: 20305825
- Ammerpohl O, Pratschke J, Schafmayer C, Haake A, Faber W, von Kampen O, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer 2012; 130:1319 - 28; http://dx.doi.org/10.1002/ijc.26136; PMID: 21500188
- Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics 2010; 283:341 - 9; http://dx.doi.org/10.1007/s00438-010-0522-y; PMID: 20165882
- Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology 2012; 55:1799 - 808; http://dx.doi.org/10.1002/hep.25569; PMID: 22234943
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 2007; 3:2023 - 36; http://dx.doi.org/10.1371/journal.pgen.0030181; PMID: 17967063
- Sandoval J, Heyn HA, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011; 3:771 - 84; http://dx.doi.org/10.2217/epi.11.105; PMID: 22126295
- Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe HH, et al. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes 2012; 5:210; http://dx.doi.org/10.1186/1756-0500-5-210; PMID: 22546179
- Beyan H, Down TA, Ramagopalan SV, Uvebrant K, Nilsson A, Holland ML, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res 2012; 22:2138 - 45; http://dx.doi.org/10.1101/gr.134304.111; PMID: 22919074
- Robinson MD, Stirzaker C, Statham AL, Coolen MW, Song JZ, Nair SS, et al. Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res 2010; 20:1719 - 29; http://dx.doi.org/10.1101/gr.110601.110; PMID: 21045081
- Jain S, Chen S, Chang KC, Lin YJ, Hu CT, Boldbaatar B, et al. Impact of the location of CpG methylation within the GSTP1 gene on its specificity as a DNA marker for hepatocellular carcinoma. PLoS One 2012; 7:e35789; http://dx.doi.org/10.1371/journal.pone.0035789; PMID: 22536438
- Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutat Res 2011; 727:55 - 61; http://dx.doi.org/10.1016/j.mrrev.2011.04.001; PMID: 21514401
- Higgs MR, Lerat H, Pawlotsky JM. Downregulation of Gadd45beta expression by hepatitis C virus leads to defective cell cycle arrest. Cancer Res 2010; 70:4901 - 11; http://dx.doi.org/10.1158/0008-5472.CAN-09-4554; PMID: 20530689
- Ahmed R, Salama H, Fouad A, Sabry D, AbdAlah S, Kamal M. Detection of aberrant p16INK4A methylation in sera of patients with HCV-related liver diseases: An Egyptian study. Med Sci Monit 2010; 16:CR410 - 5; PMID: 20802412
- Guo N, Chen R, Li Z, Liu Y, Cheng D, Zhou Q, et al. Hepatitis C virus core upregulates the methylation status of the RASSF1A promoter through regulation of SMYD3 in hilar cholangiocarcinoma cells. Acta Biochim Biophys Sin (Shanghai) 2011; 43:354 - 61; http://dx.doi.org/10.1093/abbs/gmr021; PMID: 21450690
- Lim JS, Park SH, Jang KL. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett 2012; 321:154 - 61; http://dx.doi.org/10.1016/j.canlet.2012.01.044; PMID: 22326283
- Ripoli M, Barbano R, Balsamo T, Piccoli C, Brunetti V, Coco M, et al. Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res 2011; 160:74 - 81; http://dx.doi.org/10.1016/j.virusres.2011.05.014; PMID: 21640770
- Komatsu Y, Waku T, Iwasaki N, Ono W, Yamaguchi C, Yanagisawa J. Global analysis of DNA methylation in early-stage liver fibrosis. BMC Med Genomics 2012; 5:5; http://dx.doi.org/10.1186/1755-8794-5-5; PMID: 22281153
- Mas VR, Maluf DG, Archer KJ, Yanek K, Kong X, Kulik L, et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol Med 2009; 15:85 - 94; http://dx.doi.org/10.2119/molmed.2008.00110; PMID: 19098997
- Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, et al. Gene expression patterns in human liver cancers. Mol Biol Cell 2002; 13:1929 - 39; http://dx.doi.org/10.1091/mbc.02-02-0023.; PMID: 12058060
- Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007; 45:938 - 47; http://dx.doi.org/10.1002/hep.21622; PMID: 17393520
- Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci 2010; 101:36 - 45; http://dx.doi.org/10.1111/j.1349-7006.2009.01383.x; PMID: 19891661
- Nishida N, Kudo M, Nagasaka T, Ikai I, Goel A. Characteristic pattern of DNA methylation alterations predict emergence of human hepatocellular carcinoma. Hepatology 2012; 55; http://dx.doi.org/10.1002/hep.25706
- Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol 2011; 4:30; http://dx.doi.org/10.1186/1756-8722-4-30; PMID: 21729319
- Kin T, Ono Y. Idiographica: a general-purpose web application to build idiograms on-demand for human, mouse and rat. Bioinformatics 2007; 23:2945 - 6; http://dx.doi.org/10.1093/bioinformatics/btm455; PMID: 17893084
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 2002; 99:3740 - 5; http://dx.doi.org/10.1073/pnas.052410099; PMID: 11891299