Abstract
The human activating receptor NKG2D is mainly expressed by NK, NKT, γδ T and CD8+ T cells and, under certain conditions, by CD4+ T cells. This receptor recognizes a diverse family of ligands (MICA, MICB and ULBPs 1–6) leading to the activation of effector cells and triggering the lysis of target cells. The NKG2D receptor-ligand system plays an important role in the immune response to infections, tumors, transplanted graft and autoantigens. Elucidation of the regulatory mechanisms of NKG2D is therefore essential for therapeutic purposes. In this study, we speculate whether epigenetic mechanisms, such as DNA methylation and histone acetylation, participate in NKG2D gene regulation in T lymphocytes and NK cells. DNA methylation in the NKG2D gene was observed in CD4+ T lymphocytes and T cell lines (Jurkat and HUT78), while this gene was unmethylated in NKG2D-positive cells (CD8+ T lymphocytes, NK cells and NKL cell line) and associated with high levels of histone H3 lysine 9 acetylation (H3K9Ac). Treatment with the histone acetyltransferase (HAT) inhibitor curcumin reduces H3K9Ac levels in the NKG2D gene, downregulates NKG2D transcription and leads to a marked reduction in the lytic capacity of NKG2D-mediated NKL cells. These findings suggest that differential NKG2D expression in the different cell subsets is regulated by epigenetic mechanisms and that its modulation by epigenetic treatments might provide a new strategy for treating several pathologies.
Introduction
NKG2D (natural-killer group 2, member D) belongs to the family of C-type lectin-like receptors and is encoded by the KLRK1 gene on human chromosome 12 within the NK gene complex.Citation1 NKG2D is associated with the DAP10 adaptor molecule, which is essential for signaling and the surface expression of the NKG2D receptor.Citation2 This receptor is expressed in all NK cells, most NKT, γδ T and CD8+ T cells but is not detectable in CD4+αβ+ T lymphocytes. However, an unusual CD4+ T subset expressing NKG2D (CD4+NKG2D+ T cells) was detected in subjects with tumors,Citation3 autoimmune diseases,Citation4-Citation7 persistent infectionsCitation8,Citation9 and during aging.Citation10 NKG2D is one of the most potent activating receptors of NK cells, enhancing the cytotoxic response in humans. Moreover, NKG2D can also acts as a primary or co-stimulatory receptor in CD8+ and CD4+NKG2D+ T cells, increasing the T cell receptor (TCR)-mediated signaling necessary for their activation.Citation11
Since NKG2D receptor function depends on its correct expression on the cell surface of cytotoxic cells, it is necessary to understand the factors involved in its modulation and expression. A range of factors has been implicated in the transcriptional regulation of NKG2D. Some cytokines, such us IL-2, IL-7, IL-15 and IFN-α, increase NKG2D expression, whereas IL-4, IL-12, IL-21, TGF-β and IFN-γ have the opposite effect.Citation12,Citation13 NKG2D downregulation is attributed to the overexposure to soluble or membrane-bound NKG2D ligands (NKG2DL), which promotes the internalization and subsequent degradation of the receptorCitation14 or catabolites produced on macrophage activation (reactive oxygen species and L-kynurenine).Citation15,Citation16 Additionally, the availability of the adaptor protein DAP10 is a decisive factor in NKG2D surface expression.Citation17 Recently, expression of miRNAs has been found to downregulate NKG2D expression in NK cells, damping its cytotoxic function.Citation18
Human NKG2DL, MICA, MICB (MHC class I chain-related A and B) and ULBPs 1–6 (UL-16 binding protein) are expressed at low levels in many normal tissues.Citation19,Citation20 However, their expression is induced during genotoxic or cellular stress caused by infection or malignant transformation, alerting the immune system to adverse cellular conditions. NKG2D-NKG2D ligand interactions play an important role in tumor immune surveillance.Citation21 Conversely, aberrant expression of NKG2DL in healthy cells might lead to inappropriate activation of cytotoxic NK and CD8+ T cells and trigger autoimmunity or rejection after transplantation.Citation22,Citation23 Understanding the mechanism that regulates NKG2D expression may help the development of new therapeutic strategies.
In recent years, it has been well documented that epigenetic mechanisms such as DNA methylation and histone modifications regulate the expression of key immune system-related genes, modifying the development of the immune responses.Citation24-Citation29 One advantage of epigenetic modifications is that they can be modulated by treatment with HDAC (histone deacetylase) and DNMT (DNA methyltransferase) inhibitors, some of which have already been approved by the FDA for the treatment of myelodysplastic syndromes and acute myeloid leukemia.Citation30
In this study, we show for the first time that epigenetic mechanisms regulate the differential NKG2D expression in human T- and NK-derived cell lines and in the cell subsets from peripheral blood (CD4+ T and CD8+ T lymphocytes and NK cells). We observed that DNA methylation in the NKG2D locus correlates with the absence of expression in CD4+ T cells and T cell lines (Jurkat and HUT78), whereas high levels of acetylation in lysine 9 of histone 3 (H3K9Ac) contribute to the higher level of NKG2D expression in CD8+ T cells, NK cells and the NKL cell line. Induced hypoacetylation at the NKG2D gene by curcumin,Citation31 a histone acetyltransferase (HAT) inhibitor, downregulates NKG2D-receptor expression on the cell surface of cytotoxic cells, damaging its lytic activity. Modulation of NKG2D receptor by epigenetic modifiers could represent a significant area of interest with important implications in the improvement of new therapies targeting NKG2D.
Results
NKG2D expression in T cell lines and CD4+ T lymphocytes is regulated by DNA methylation
We had previously determined the expression of NKG2D receptor on the cell surface of T (Jurkat and HUT78) and NK (NKL) cell lines and cell subsets of peripheral blood (CD4+ T, CD8+ T and NK cells). As has been previously reported,Citation32 we observed that receptor is expressed in all CD8+, NK and NKL cells but remains absent in Jurkat and HUT78 cell lines (both derived from CD4+ T cells) and CD4+ T lymphocytes (Fig. S1A). We also examined whether the absence of NKG2D expression might be caused by the lack of transcription of the NKG2D gene or its DAP10 co-receptor. To do this, NKG2D and DAP10 transcripts were quantified by quantitative real-time reverse transcription PCR (qRT-PCR) in the same cell samples. As expected, NKG2D transcripts were only detected in CD8+ T, NK and NKL cells () while DAP10 expression was observed in all the subsets analyzed although at different levels of expression (Fig. S1B). These findings suggest that the lack of NKG2D expression on the cell surface of CD4+ T lymphocytes and T cell lines might be due to the absence of transcripts for NKG2D gene expression but not of its adaptor molecule DAP10, which is expressed in all cell types.
Figure 1. Differential expression of NKG2D gene in T and NK cell lines and lymphocyte subsets is correlated with DNA methylation pattern.(A) NKG2D transcription levels were determined by qRT-PCR in Jurkat, HUT78 and NKL cell lines, and isolated CD4+ T, CD8+ T and NK cells from healthy donors. Histograms represent the relative expression of NKG2D gene normalized with respect to the housekeeping gene GAPDH. Data are presented as the mean ± SD of five independent experiments. (B) Graphic representation of the NKG2D gene. Arrow: translation start site; filled bars: distribution and position of CpG sites; horizontal black lines: regions amplified with specific primers shown in . The methylation pattern of each cell type was analyzed by bisulfite sequencing. Rows: ten individual sequenced clones; columns: CpG sites; closed squares: methylated CpG sites; open squares: unmethylated CpG sites. The right-hand graphs show the total percentage methylation for each sample.
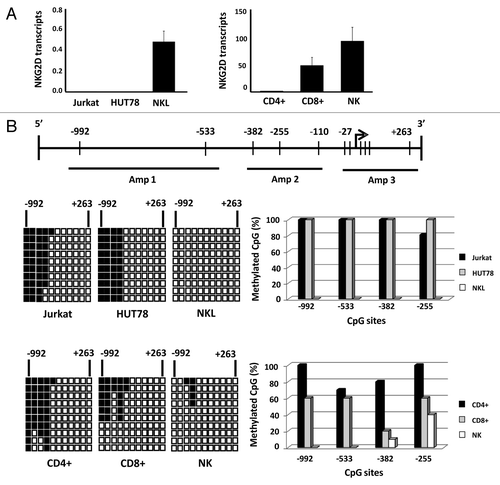
In view of these results, we determined whether epigenetic mechanisms such as DNA methylation could be involved in regulating the NKG2D gene in these cell subsets. A region located around the translation initiation site between -992 and +263 positions and including 11 CpG sites was analyzed by bisulfite sequencing (). The greatest differences in the DNA methylation patterns were found in a region localized between the positions -992 and -255. These CpGs were highly methylated (80–100%) in Jurkat and HUT78 cell lines, and fully demethylated in NKL cells. Similarly, these CpG sites were highly methylated (70–100%) in CD4+ T cells, but only partially methylated (10–60%) in CD8+ T lymphocytes and NK cells. These results demonstrate that the DNA methylation pattern is correlated with the expression of NKG2D gene, suggesting that hypermethylation of this region could be involved in the lack of expression of NKG2D in T cell lines and CD4+ T lymphocytes. The DNA methylation profile in DAP10 gene was also analyzed, but no differences were found (data not shown).
To investigate whether DNA demethylation of this region is sufficient for the correct NKG2D transcription, we treated Jurkat and CD4+ T cells with the DNMT inhibitors, 5azaC (5-azacytidine) and DAC (5-aza-2'deoxycytidine), and NKG2D expression was assayed by qRT-PCR. Previously, we determined that the analyzed concentrations of DNMT inhibitors do not compromise cell viability (Fig. S2). Our results showed that treatment with both inhibitors increased significantly the transcription level of NKG2D in a time-dependent manner in the Jurkat cell line but expression on the cell surface was not detected (). Jurkat cells were more sensitive to the action of DAC than 5azaC-treated cells and the highest level of NKG2D transcription was observed at 72 h. Additionally, CD4+ T lymphocytes were activated with anti-CD3 and anti-CD28 monoclonal antibodies (mAb) over four days and further treated with 5azaC and DAC. Treatment with both demethylating agents induce a significantly NKG2D transcription in CD4+ T cells and a slight proportion of NKG2D-positive CD4+ T cells was observed by flow cytometry ().
Figure 2. Effect of DNMT inhibitors (5azaC and DAC) in NKG2D expression in Jurkat and CD4+ T cells. (A) Jurkat cells were treated with 5azaC (1–5 μM) and DAC (60–80 μM) for 24, 48 and 72 h and NKG2D transcripts were quantified by qRT-PCR using specific primers (). Histograms show the fold increase of NKG2D expression after treatment relative to untreated cells. GAPDH was used as a constitutive control and data are presented as the mean ± SD of three independent experiments. The right panel shows representative histograms of NKG2D expression on the cell surface of Jurkat cells after DNMT inhibitors treatment for 72 h. (B) NKG2D transcription levels were determined by qRT-PCR in CD4+ T lymphocytes from healthy donors. Isolated CD4+ cells were previously activated with anti-CD3 and anti-CD28 mAb over four days, and further cultured in the absence or presence of DAC (1 μM) and 5azaC (5 μM) for 72 h. The histogram shows the fold increase of NKG2D expression after treatment relative to untreated cells. GAPDH was used as a constitutive control for qRT-PCR. Data are presented as the mean ± SD of three independent experiments. The right panel shows representative dot plots of the induced NKG2D expression on CD4+ T cells after DNMT inhibitor treatment. The number indicates the percentage of NKG2D-positive CD4+ T cells. *p < 0.05. Significance is shown as the difference between treated and untreated Jurkat or CD4+ T cells.
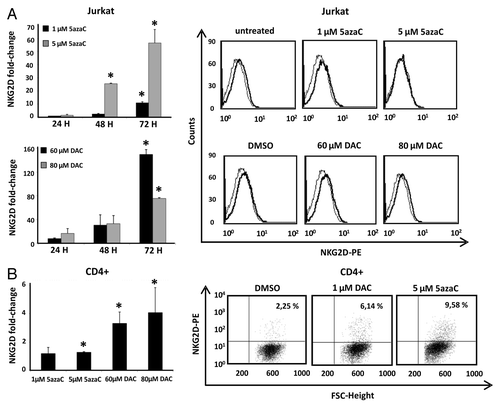
Taken together, these results suggest that DNA methylation contributes to the regulation of NKG2D expression, although additional mechanisms may be involved, as revealed by the low level or complete absence of expression of NKG2D receptor on the cell surface of CD4+ T lymphocytes and Jurkat cells after treatment with demethylating agents.
Acetylation of H3K9 is associated with an active transcription of the NKG2D gene in CD8+ T and NK cells
Chromatin structure is controlled by posttranslational modification of the N-terminal histone tail regions, which are associated with gene activation or silencing. Here, we analyzed the distribution of active and repressive histone modifications in a region comprised of 382 base pairs upstream of the translation initiation site of the NKG2D gene from human cell lines (Jurkat, HUT78, and NKL) () and CD4+ and CD8+ T lymphocytes and NK cells (). Two pairs of primers were designed to cover the full region ().
Figure 3. Enrichment of active and repressive histone modifications in NKG2D gene. Levels of histone modifications in NKG2D gene were measured by chromatin immunoprecipitation (ChIP) assay using antibodies specific for AcH4, AcH3, H3K9Ac, H3K4me, H3K27me3, H3K9 me2 in the Jurkat, HUT78 and NKL cell lines (A) and in CD4+, CD8+ T lymphocytes and NK cells (B). Normal rabbit IgG was used as a negative control. Enrichment of NKG2D-specific DNA sequences was measured by quantitative PCR with specific primers (). Two consecutive regions in the NKG2D gene were amplified and the average of both products is shown. Results are represented as % INPUT = 2 exp [Ct (BOUND) – (Ct (UNBOUND) - log2 (UNBOUND DILUTION FACTOR))] × 100. Data are shown from two independent experiments and each qRT-PCR was performed in triplicate.*p < 0.05 for comparisons of Jurkat and NKL cells, and of CD4+ T cells and NK cells. #p < 0.05 for comparisons of HUT78 and NKL cells, and of CD4+ T with CD8+ T cells.
![Figure 3. Enrichment of active and repressive histone modifications in NKG2D gene. Levels of histone modifications in NKG2D gene were measured by chromatin immunoprecipitation (ChIP) assay using antibodies specific for AcH4, AcH3, H3K9Ac, H3K4me, H3K27me3, H3K9 me2 in the Jurkat, HUT78 and NKL cell lines (A) and in CD4+, CD8+ T lymphocytes and NK cells (B). Normal rabbit IgG was used as a negative control. Enrichment of NKG2D-specific DNA sequences was measured by quantitative PCR with specific primers (Table 1). Two consecutive regions in the NKG2D gene were amplified and the average of both products is shown. Results are represented as % INPUT = 2 exp [Ct (BOUND) – (Ct (UNBOUND) - log2 (UNBOUND DILUTION FACTOR))] × 100. Data are shown from two independent experiments and each qRT-PCR was performed in triplicate.*p < 0.05 for comparisons of Jurkat and NKL cells, and of CD4+ T cells and NK cells. #p < 0.05 for comparisons of HUT78 and NKL cells, and of CD4+ T with CD8+ T cells.](/cms/asset/0a0fa1d6-7ce5-412c-a3bd-b69e7fc3fcc8/kepi_a_10923115_f0003.gif)
Table 1. PCR and qRT-PCR primers
First, we analyzed the presence of active histone modifications (AcH3, AcH4, H3K9Ac and H3K4me3) by ChIP assay and further amplification by quantitative PCR. Levels of acetylation of histone 3 (AcH3) and histone 4 (AcH4) were increased in the NKG2D gene in the NKL cell line but absent from the Jurkat and HUT78 cell lines, the levels of AcH3 being higher than those of AcH4 (). Similar results were obtained in isolated CD4+ T, CD8+ T and NK cells from peripheral blood, in which a significant increase in AcH3 in CD8+ T lymphocytes and NK cells was observed (). In view of these results we investigated the presence of acetylation at a specific residue of H3: lysine 9 (H3K9Ac). A significant increase in the H3K9Ac level of the NKG2D gene was detected in CD8+ T, NK and NKL cells. The presence of these active histone marks was correlated with NKG2D expression in these cell subsets. Another histone modification, lysine 4 trimethylation of H3 (H3K4me3), was significantly more prevalent in NKL cells than in other T cell lines (). However, low and similar levels of H3K4me3 were present in all subset cells isolated from peripheral blood (CD4+ T and CD8+ T and NK cells) (), suggesting its lack of involvement in NKG2D regulation.
We also assayed the contribution of repressive histone modifications (H3K27me3 and H3K9me2) to NKG2D transcription (). These marks were hardly detectable in the NKG2D gene in comparison with the active marks previously analyzed, showing their slighter contribution to the regulation of NKG2D expression. Thus, active transcription of NKG2D in CD8+ T, NK and NKL cells is characterized by high levels of H3K9Ac and reduced levels of repressive marks, which is consistent with the current paradigm of gene activation.
Induced deacetylation of H3K9 downregulates NKG2D expression
To verify whether the H3K9Ac level is specifically involved in regulating NKG2D expression in the different cell types, we treated cells with HDAC and HAT inhibitors. Jurkat cells were cultured with the HDAC inhibitor, valproic acid (VPA), at concentrations that do not compromise the cell viability and, in the absence or presence of IL-15 cytokine (; Fig. S3A). This cytokine is known to upregulate NKG2D expression on the cell surface of CD4+ T lymphocytes.Citation7 A dose-dependent and significant increase in NKG2D expression was observed in Jurkat cells, and it had an additive effect in combination with IL-15.
Figure 4. Effect of HDAC and HAT inhibitors in NKG2D expression in Jurkat and NKL cell lines. (A) NKG2D expression was measured in the Jurkat cell line after treatment with VPA (0.5–1.0 mM) in the absence or presence of IL-15 (50 ng/ml) cytokine. Cells were harvested after four days and NKG2D transcripts were quantified by qRT-PCR using specific primers (). The histogram shows the fold increase in NKG2D expression after treatment relative to untreated cells. GAPDH was used as a constitutive control for qRT-PCR. Data are represented as mean ± SD of three independent experiments *p < 0.05. Significance is shown as the difference between untreated and treated Jurkat-cells. (B) NKG2D expression in NKL cells was analyzed before and after treatment with 4 μM curcumin for 24 h by flow cytometry. The histogram is representative of five independent experiments. (C) NKG2D and DAP10 transcripts were quantified by qRT-PCR in untreated and curcumin-treated NKL cells (4 µM, 24 h). GAPDH was used as a control and the results are represented as the mean ± SD of three independent experiments. (D) The level of H3K9Ac in the NKG2D gene was analyzed in untreated and curcumin-treated NKL cells by ChIP assay. Normal rabbit IgG was used as a negative control. Data are expressed as the % INPUT from three independent experiments and qRT-PCR was performed in triplicate. *p < 0.05
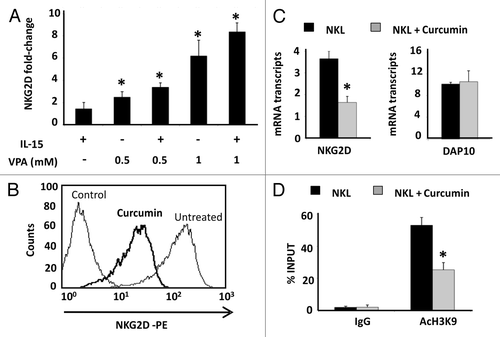
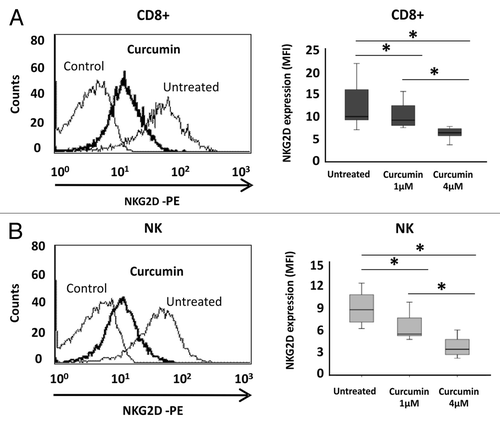
We also used a HAT inhibitor, curcumin, to induce hypoacetylation and analyzed how the NKG2D expression was modified. Interestingly, NKG2D expression was significantly reduced [6.04-fold ± 0.13 of Mean Fluorescence Intensity (MFI) ratio; p < 0.05] on the cell surface of NKL cells after treatment with 4 μM curcumin for 24 h (). Downregulation of NKG2D receptor was associated with decreased transcription of the NKG2D gene (2.23-fold ± 0.23, p < 0.05) but the levels of the DAP10 co-receptor remained unchanged (). To confirm that decreased NKG2D expression after curcumin-treatment is due to histone deacetylation, we determined the levels of H3K9Ac in the NKG2D gene in the NKL cell line after treatment (4 μM curcumin) by ChIP assay. Strikingly, a significantly reduction in H3K9Ac levels at the NKG2D locus in curcumin-treated NKL cells compared with untreated NKL cells was observed (). Similar results were observed in isolated CD8+ T and NK cells from healthy donors after treatment with curcumin (1 and 4 μM) for 24 h. We observed a significant and dose-dependent downregulation of NKG2D expression on the cell surface of both cell types (). The NKG2D expression in curcumin-treated cells showed a 2.71-fold ± 1.08 and 2.98-fold ± 1.03; p < 0.05 decrease with respect to untreated cells at the highest analyzed concentration in CD8+ T and NK cells, respectively.
Figure 5. Downregulation of NKG2D expression in CD8+ T lymphocytes and NK cells after curcumin treatment. NKG2D expression on the cell surface of CD8+ T cells (A) and NK cells (B) was assayed before and after treatment with curcumin (1–4 μM) for 24 h by flow cytometry. Left-hand panels show a representative histogram after treatment with 4 μM curcumin for 24 h; on the right, box-plots indicate NKG2D expression in CD8+ T lymphocytes and NK cells from ten healthy donors before and after curcumin treatment (1–4 μM). Data are expressed as the Mean Fluorescence Intensity (MFI) of NKG2D expression. *p < 0.05.
Additionally, we analyzed the influence of curcumin in the cell viability and proliferation of these cells by 7AAD/Annexin V staining and MTT assay. In both cell types (NKL and PBLs) the concentrations used in our experiments (1 and 4 µM) not inhibit cell proliferation showing cell viability above 88% (Fig. S3B and C).
Taken together, these results suggest that the H3K9Ac levels in the NKG2D gene might contribute to the expression of NKG2D receptor on the cell surface of cytotoxic CD8+ T and NK cells.
Curcumin reduces NKG2D-dependent NKL cell line cytotoxicity
As it had been previously reported, NKG2D is one of the main activating receptors expressed on NK cells.Citation2 The NKG2D-NKG2D engagement activates the effector functions of cytotoxic cells, leading to the release of cytotoxic granules and to the lysis of target cells. Since curcumin downregulates the NKG2D-receptor expression, we performed cytotoxicity assays to study the effect of this drug on the lytic capacity of NKG2D-dependent NKL cells. We did this using a well-known model that allowed the analysis of the NKG2D-MICA interaction in a specific way. As effector cells, we used the NKL cell line, which expresses high levels of NKG2D receptor (Fig. S1A) and is therefore very suitable for studying NKG2D-mediated recognition. As target cells, we used the C1R class I-negative cell line stably transfected with the NKG2DL MICA*004 (C1R-MICA*004), as we previously reported.Citation33 As control cells, we used the C1R cell line transfected with the vector alone.
First, we confirmed that the lytic activity of NKL cells was significantly higher in the CIR-MICA*004 transfectants than in the control C1R cell line for all effector:target (E:T) ratios analyzed (). When NKL cells were previously incubated with anti-NKG2D blocking mAb for 30 min, the percentage of specific lysis was significantly lower in all assayed E:T ratios.
Figure 6. Curcumin suppresses NKG2D-mediated NKL cell line cytotoxicity. (A) NKL cells were cultured with C1R cells and C1R-MICA*004 transfectants at the indicated effector:target (E:T) ratios for 4 h. Cytotoxicity was assayed by flow cytometry after PKH67/7AAD staining. For blocking assays, NKL cells were incubated beforehand with anti-NKG2D blocking monoclonal antibody (10 μg/ml) for 30 min. Data are shown as means ± SD of two independent experiments in triplicate. (B) NKL cells were incubated in the absence or presence of curcumin (4 µM for 24 h) and cytolytic activity against C1R and C1R-MICA*004 cells was assessed at the indicated effector: target (E:T) ratios. The upper figure shows a representative experiment and the lower histogram represents means ± SD of three independent experiments performed in triplicate. *p < 0.05 comparison between untreated and curcumin- or anti-NKG2D-treated NKL cells against C1R-MICA*004 transfectants. #p < 0.05 C1R-MICA*004 compared with C1R cells.
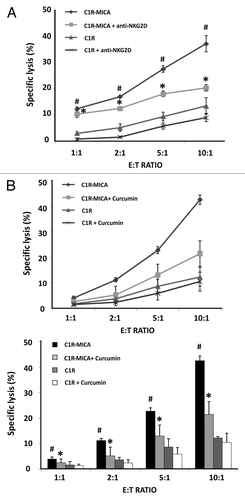
We next assayed the effect of NKG2D downregulation after curcumin treatment on the cytolytic activity of NKL cells. To do this, untreated and curcumin-treated NKL cells were co-cultured for 4 h with target cells at the indicated E:T ratios. shows the percentage of specific lysis of C1R-MICA*004 cells and C1R cells for untreated and curcumin-treated NKL cells. Interestingly, the cytotoxicity of curcumin-treated NKL cells against C1R-MICA*004 was significantly reduced in an E:T ratio-dependent manner.
These findings show that curcumin, a HAT inhibitor, inhibits NKG2D-mediated NKL cell cytotoxicity by suppressing NKG2D activation receptor expression. Treatments with epigenetic modifiers might be useful for controlling the effector functions of cytotoxic cells.
Curcumin damages NKG2D-dependent NK and CD8+ T cell degranulation and INF- γ production by NKL cells
After recognition of target cells, NK cells release cytolytic granules containing granzyme B (GZMB) and perforin (PRF1), and produce IFN-γ cytokine, thereby contributing to the cytolysis of target cells. To determine whether curcumin modulates other components of the cytolytic machinery in addition to downregulating NKG2D, we quantified the CD107a degranulation in CD8+ T and NK cells from peripheral blood and the intracellular IFN-γ production by NKL cells by flow cytometry. Unfortunately, degranulation assays were not done in the NKL cell line due to high expression levels of CD107a under basal conditions.
Previous studies had shown that curcumin inhibits the release of granzyme B and perforin in CD8+ T lymphocytes.Citation34 In this study, we determined the effect of curcumin on NKG2D-mediated CD8+ T and NK cell degranulation. CD107a expression was analyzed after incubation of these cells with anti-NKG2D-activating mAb-coated plates for 6 h. Impaired degranulation (CD107a expression) was observed in both cell types upon treatment with curcumin (). Additionally, we observed that the level of expression of granzyme B and perforin in NKL cells dropped markedly after curcumin treatment, showing a decrease of 2.15-fold ± 0.18 and 2.14-fold ± 0.73; p < 0.05 changes in PRF1 and GZMB transcripts, respectively. This downregulation was also observed by intracellular staining with specific antibodies (). Further, NKL cells previously treated or not with curcumin for 24 h were incubated with C1R and C1R-MICA*004 target cells, and IFN-γ production was assayed by intracellular staining. According to the observed effects on the cytotoxicity, curcumin-treated cells showed impaired IFN-γ production in the presence of C1R-MICA*004 compared with untreated NKL cells ().
Figure 7. Curcumin modifies the cytolytic machinery in CD8+ T lymphocytes, NK and NKL cells. (A) Frequency of CD107a+ degranulation by T CD8+ and NK cells after stimulation with anti-NKG2D mAb. Untreated or curcumin-treated (4 µM) CD8+ and NK cells were cultured for 6 h in the presence of activating anti-NKG2D mAb or isotype control, and expression of membrane CD107a was measured by flow cytometry. Data represent the mean ± SD values from triplicate experiments. (B) Granzyme B and perforin expression was analyzed in the NKL cell line before and after treatment with 4 µM of curcumin for 24 h by qRT-PCR and flow cytometry. GAPDH was used as a control and results are represented as the mean ± SD of three independent experiments. Histograms are representative of an experiment for intracellular staining of granzyme B and perforin. (C) Frequency of IFN-γ production by untreated or 4 µM curcumin-treated NKL cells after coculture for 6 h with C1R or C1R-MICA cells at E: T ratio 1:1. Data represent mean ± SD values from triplicate experiments.*p < 0.05.
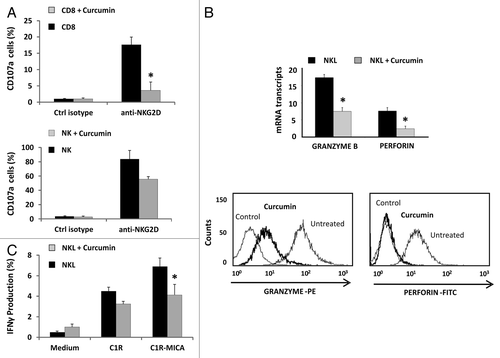
These results imply that curcumin treatment not only inhibits NKG2D expression but also impairs the production of other molecules essential for the lytic activity of cytotoxic CD8+ T and NK cells.
Discussion
The NKG2D activating receptor functions as a sensory signal that alerts the immune system to adverse cellular conditions, such as infections and tumors. However, NKG2D can also recognize NKG2DL induced on healthy target cells, thereby contributing to the development of autoimmune diseases and transplant rejection. Thus, the knowledge of the mechanisms involved in the expression and function of NKG2D could be of great relevance to the development of new therapeutic strategies for treating these pathological conditions.
In the present study, we have shown that epigenetic mechanisms such as DNA methylation and histone modifications contribute to the regulation of NKG2D expression in the diverse cell subsets from peripheral blood. We have also demonstrated that the reduced levels of H3K9Ac in NKG2D gene caused by a HAT inhibitor (curcumin) decrease NKG2D expression and damp the cytotoxic capacity of NK cells. The modulation of NKG2D expression through epigenetic modifiers might have important consequences for the function of cytotoxic cells.
Differences in the DNA methylation pattern in the NKG2D gene were observed among CD4+, CD8+ T lymphocytes and NK cells. As had been reported previously, the NKG2D receptor is expressed in a constitutive form in NK and CD8+ T cells but not in CD4+ T lymphocytes.Citation32 Our results reveal DNA hypermethylation in the NKG2D gene in Jurkat and HUT78 cells lines and in CD4+ T lymphocytes. However, partial or full demethylation was observed in the cell types that express NKG2D (CD8+ T, NK and NKL cells). These findings suggest that high levels of DNA methylation in the NKG2D gene may contribute to NKG2D repression in CD4+ T cells. However, additional regulatory mechanisms such as miRNA or low levels of DAP10 might be required for total NKG2D expression.
The NKG2D receptor is expressed in a certain subset of CD4+ T lymphocytes under conditions of chronic antigenic stimulation. These cells form a subset of highly differentiated T cells that lose the CD28 co-stimulatory molecule and increase the expression of NK cell-related genes (KIR, granzyme B and perforin). Some authors have suggested that the enhanced expression of these genes is a consequence of a demethylation process arising from antigen-stimulated proliferation or homeostatic turnover.Citation35,Citation36 In our study, we showed that treatment with the DNMT inhibitors, 5azaC and DAC, induces NKG2D transcription and a small proportion of CD4+ T lymphocytes express this receptor on the cell surface. Thus, these findings show that demethylation could be involved in the increased NKG2D expression in these particular CD4+ T cells.
Histone modification and DNA methylation are related and dynamic events that establish an epigenetic code ensuring the correct control of gene expression. When the presence of active and repressive histone modifications was examined, we observed high levels of H3K9Ac in NKG2D gene of CD8+ T, NK and NKL cells, but was absent from NKG2D-deficient cells. Hyperacetylation of histones at specific lysine residues neutralize the positive charge of histone tails, decreasing their affinity for negatively charged DNA, and favoring an open chromatin structure and active transcription. Thus, the presence of H3K9Ac could allow the recruitment of transcriptional factors and RNAP II to the gene locus, facilitating the continued expression of NKG2D in cytotoxic T and NK cells and maintaining their functionality. This active histone mark has been associated with the expression of the EOMES transcription factor and the perforin and granzyme B effector molecules, facilitating a rapid and robust response of memory CD8+ T cells to the re-encounter with the antigen.Citation34 Therefore, controlling the level of histone acetylation might be a promising strategy for modulating the expression of genes associated with the cytotoxic functions of the immune system.
The level of histone acetylation is established and maintained by the interplay between HATs and HDACs. HATs modulate gene expression by catalyzing the acetylation of lysine residues on histone and non-histone proteins and enhancing the transcriptional activity. Balasubramanyan et al.Citation37 reported that curcumin, a specific inhibitor of p300/CBP HAT activity, can induce histone hypoacetylation and downregulate gene expression. Based on these findings, we observed that NKG2D expression was significantly reduced in NKL cells after curcumin treatment, and was correlated with reduced levels of H3K9Ac in the NKG2D gene. Additionally, downregulation of the NKG2D receptor expression was found in ex vivo CD8+ T lymphocytes and NK cells from healthy controls after curcumin treatment. Taken together, these results suggest that H3K9Ac is tightly linked to NKG2D expression, although further studies are needed to determine the involved mechanism.
In recent years, several studies have indicated that treatment with DNMT and HDAC inhibitors can modulate the lytic capacity of NK cells. Azacytidine impairs NK cell function by overexpression of inhibitory KIR receptors, whereas decitabine enhances IFN-γ production and NK cell-mediated cytotoxicity.Citation38,Citation39 Similarly, treatments with HDAC inhibitors (VPA and SAHA) inhibit NK cell cytotoxicity by reducing expression of activating receptor Nkp30 and Nkp46 as well as impairing granule exocytosis.Citation40 However, the modulation of the NKG2D-mediated NK cell cytotoxicity has not been analyzed in depth.
To analyze the effect of NKG2D downregulation mediated by curcumin in the lytic capacity of cytotoxic cells we performed cytotoxicity assays with the NKL effector cell line, which expresses high levels of NKG2D receptor. Previous studies by our group have shown that NKL cell cytotoxicity against the C1R-MICA*004 transfectant is specific to the NKG2D-MICA interaction.Citation33 When curcumin-treated NKL cells were assayed against C1R-MICA target cells, the lytic ability of NKL effector cells was reduced significantly relative to untreated cells. Thus, downregulation of NKG2D expression by curcumin could have important functional consequences in the lytic capacity of NK cells and avoid the lysis of target cells.
NK cell cytotoxicity is a complex process that requires adhesion to target cells, synapse formation and signal transduction leading to release of cytotoxic granules and IFN-γ production. We have shown a decrease in the granzyme B and perforin expression in curcumin-treated NKL cells, as well as impaired IFN-γ production in the presence of C1R-MICA*004 target cells. In addition, degranulation of curcumin-treated CD8+ T and NK cells was decreased respect to untreated cells after incubation with anti-NKG2D mAb. In consequence, NKG2D downregulation by curcumin reduces the recognition of NKG2DL-expressing target cells, at the same time impairing the cytolytic machinery and inhibiting the release of cytolytic granules and cytokine production. Thus, the regulation of NKG2D expression by epigenetic treatments could have important functional consequences for the lytic capacity of NK cells and for preventing the elimination of target cells.
These findings could be of considerable clinical relevance in the treatment of diseases in which cytotoxic cells play an important role. The overexpression of NKG2DL in healthy cells can disrupt the balance between immune activation and tolerance, and promote autoimmune responses favoring the development of rheumatoid arthritis (RA), multiple sclerosis and celiac disease.Citation4,Citation41,Citation42 The pathogenesis of RA is characterized by excessive production of IL-15 and TNF-α in the sera, and inflamed synovial joints, which induce the expression of an unusual subset of NKG2D-expressing CD4+ T cells. Moreover, NKG2DL MICA and MICB are highly upregulated in synovial tissue of RA patients, activating the T cells in an NKG2D-dependent manner. In multiple sclerosis, the concomitant presence of IL-15-expressing astrocytes and NKG2DL-expressing oligodendrocytes may enhance the NKG2D-mediated effector functions of CD8+ T lymphocyte and consequently exacerbate the killing of oligodendrocytes.Citation41 Celiac disease is a gluten-induced intestinal inflammatory disorder that results in epithelial cell destruction in the small intestine. The associated inflammation results in elevated levels of IL-15, which induce the expression of NKG2D in intraepithelial γδT lymphocytes. Additionally, MICA and MICB proteins are markedly upregulated in patients with active disease, favoring the cytotoxicity to epithelial cells.Citation11,Citation42
The NKG2D receptor also participates in the immune responses that occur after solid organ transplantation and promote graft injury or rejection.Citation23,Citation43-Citation45 Under conditions of ischemia/reperfusion injury or rejection, NKG2DL are upregulated, favoring the interaction with its receptor and enhancing effector immune responses. Downregulation or blocking of the NKG2D receptor could also have an important impact in the case of graft-vs.-host disease (GVHD), which is a major late complication in patients treated by allogeneic hematopoietic stem cell transplantation. An induction of NKG2DL in the skin, gut and liver during GVHD has been observed. The engagement with the NKG2D receptor activates NK cells and γδ CD8+ T cells, boosting the immune response that leads to tissue damage during GVHD.Citation46
Previous results had demonstrated that targeting the NKG2D-NKG2DL interaction might help prevent these pathologies. Blocking NKG2D with neutralizing anti-NKG2D monoclonal antibodies prevents the development of autoimmune diseasesCitation47-Citation49 and increases the survival of skin, bone marrow and cardiac allograft in animal models.Citation43,Citation50,Citation51 Therefore, modulation or downregulation of NKG2D by epigenetic mechanisms could be an additional therapeutic strategy for NKG2D blockage in these pathologies.
In summary, this study has shown for the first time that epigenetic mechanisms, such as DNA methylation and histone modifications, are involved in the regulation of NKG2D expression. Acetylation of H3K9 is important for correct NKG2D expression in NK and CD8+ T cells, while DNA demethylation in the NKG2D gene may be associated with an increased expression of NKG2D in CD4+ T cells. In this paper, we propose that downregulation of NKG2D in NK and CD8+ T cells by HAT inhibitors, such as curcumin, can damp the recognition of NKG2DL-positive target cells and avoid exacerbating NKG2D-mediated cytotoxic responses in autoimmune diseases, transplant rejection or reducing the incidence of GVHD. The findings of this study may help develop new NKG2D-specific therapeutic strategies in a wide range of diseases.
Materials and Methods
Cells
Jurkat, HUT78 (both derived from CD4+ T lymphocytes), NKL (derived from NK cells) and C1R (human B-cell lymphoma) cell lines were grown in RPMI-1640 medium plus 10% heat-inactivated fetal bovine serum (FBS) and 100 U/ml streptomycin-penicillin. The NKL cell line was supplemented with 100 U/IL-2 (Prepotech). C1R-MICA*004 transfectants were maintained in complete medium supplemented with hygromycin B at 800 µg/ml (Invitrogen). PBLs were obtained from healthy donors by Ficoll (Lymphoprep) density gradient centrifugation after obtaining informed consent in accordance with the Declaration of Helsinki. CD4+, CD8+ T cells and NK cells were enriched from PBLs by negative selection using magnetic microbeads (Invitrogen, CA). The purity of isolated cells was always > 95%.
Flow cytometry analysis
Surface expression of NKG2D was analyzed with a FACSCalibur flow cytometer (BD Biosciences) using the following mAbs: PE-anti-NKG2D, APC-anti-CD4, PerCP-anti-CD8, FITC-anti-CD56, APC-anti-CD16 and PerCP-anti-CD3 and isotype-matched mAb (negative control for nonspecific binding) (all from eBiolegend). For intracellular staining of cytotoxic molecules, cells were fixed and permeabilized with a Fixation/Permeabilization kit (Immunostep Inc.) and stained with anti-granzyme B-PE or anti-perforin-FITC for 30 min at room temperature. The proportions of living, dead and apoptotic cells were determined with 7AAD and the Annexin-V-FITC apoptosis detection kit (Immunostep Inc.).
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA was isolated using the RNeasy® Plus Mini Kit (Qiagen) and 1 μg was reverse-transcribed with an iScriptTM cDNA Synthesis kit (Bio-Rad). Quantitative real-time PCR was performed in triplicate with the MyiQ™ Real Time PCR Detection System (Bio-Rad) using iQTM SYBR® Green Super Mix (Bio-Rad). The numbers of transcripts were calculated using threshold cycle (Ct) values standardized to GAPDH, applying the 2-(ΔCt) method. Specific primers () were designed with the Primer3 program.Citation52
Bisulfite modification and DNA sequencing
Genomic DNA was isolated using the DNeasy® Blood and Tissue kit (Qiagen) and modified with bisulfate using the EZ-DNA Methylation-Gold™ kit (Zymo Research), according to the manufacturers’ instructions. DNA was amplified using specific primers designed with the Methyl Primer Express® program (Applied Biosystems) (). PCR products were cloned into the pGEM® Easy Vector System II (Promega) and ten independent clones from each sample were sequenced.
Treatments with DNMT, HDAC and HAT inhibitors
Jurkat and HUT78 cell lines were cultured at a density of 0.5 × 106 cells/ml in six-well plates and treated with the DNMT inhibitors, 5azaC (1–5 μM) and DAC (60–80 μM) (Sigma-Aldrich) at the indicated concentrations for 24, 48 and 72 h. PBLs (106 cells/well) were cultured for 4 d in 24-well plates previously coated with anti-CD3 (1 µg/ml) and anti-CD28 (0.5 μg/ml) overnight at 4°C. After that, cells were treated with 5azaC (5 μM) and DAC (1 μM) for a further 72 h. Jurkat cells were cultured at a density of 0.5 x 106 cells/ml in six-well plates and treated with VPA (0.5–1mM) (Sigma-Aldrich) for 4 d in the presence or absence of 50 ng/ml IL-15 cytokine (eBiosciences, San Diego, CA, USA). The NKL cell line (106 cells/ml) and PBLs (106 cells/ml) were cultured with 4 μM of curcumin (Sigma-Aldrich) for 24 h.
Cell proliferation assay
For the proliferation assay, NKL cells and PBLs (104 cells/100 µl) were plated onto 96-well plates in complete RPMI 1640 medium containing IL-2 (100 U/ml) at 37°C in a 5% CO2 incubator. After 24 h, cell proliferation was assayed by the MTT colorimetric method. MTT (3–2, 5-diphenyltetrazolium bromide; Sigma) was added at 5 mg/ml to all wells, and plates were incubated for 4 h at 37°C. After that, cell supernatant was removed and DMSO (100 µl/well) was added to dissolve MTT-formazan crystals. Absorbance at 595 nm was determined with an automated microtiter plate reader. Optical density was directly proportional to cell number up to the maximum density measured.
Cytotoxicity assay
Cytotoxicity was assayed using the PKH67 Green Fluorescent Cell Linker kit (Sigma-Aldrich) and by flow cytometry, as previously described.Citation53 NKL cells, previously treated or not with curcumin (4 μM, 24 h) were tested for cytotoxicity against C1R and C1R-MICA*004 target cells at different effector: target (E:T) ratios. After incubation at 37°C for 4 h, cells were stained with 5 µl 7AAD and analyzed by flow cytometry. Target cells incubated in the absence of effector cells were used as a negative control. Percentage specific lysis was calculated using the formula: % NK-specific lysis = 100 × [(% 7AAD staining of sample – % 7AAD staining of negative control) / (100 – % 7AAD staining of negative control)]. For blocking experiments, NKL cells were incubated beforehand with 10 μg/ml anti-human NKG2D blocking monoclonal antibody (eBioscience) for 30 min.
Degranulation assay and INF-γ production
PBLs (1 × 106 cells/ml) were treated or not with 4 µM of curcumin and incubated further on coated plates with 10 µg/ml of anti-NKG2D activating monoclonal antibody (eBiosciences) or isotype control for 6 h. CD107a-FITC antibody was added directly to the wells and after 1 h at 37°C in 5% CO2, brefeldin A (10 µg/ml, eBiosciences) and monesin (6 µg/ml, eBiosciences) were added for an additional 5 h, and cells were stained with CD8-PE, CD3-PerCp and CD16-APC antibodies (all from Biolegend), fixed and analyzed by flow cytometry.
For intracellular IFN-γ analysis, NKL cells were treated with curcumin (4 µM) or untreated and further incubated in the presence of C1R or C1R-MICA target cells at a effector:target ratio of 1:1. After 1 h, brefeldin A (10 µg/ml, eBiosciences) was added for an additional 5 h, cell were stained with anti-CD16-APC, fixed and permeabilized for intracellular staining with anti-INFγ-FITC antibody for 30 min. Cells were washed and analyzed by flow cytometry.
ChIP assay
ChIP assays were performed as previously described,Citation54 using the following antibodies: anti-acetyl-H3 (catalog number 07-352), anti-acetyl-H4 (06-598), anti-acetyl-H3K9 (07-352), anti-trimethyl-H3K4 (07-473), anti-dimethyl-H3-K9 (17-681) and anti-trimethyl-H3-K27 (07-449) antibodies. Normal rabbit IgG was used as a negative control. All antibodies were obtained from Upstate Biotechnologies Inc. In brief, fixed cells with 1% formaldehyde (Sigma-Aldrich) were lysed in SDS lysis buffer and sonicated. The shared chromatin was diluted into ChIP dilution buffer and incubated with 5 μg of each antibody overnight at 4°C. Antibody-chromatin complexes (bound sample) were precipitated from non-specific chromatin (unbound sample) with protein A-agarose/salmon sperm DNA (Upstate Biotechnologies) for 1 h, then washed and eluted from the beads with elution buffer. After cross-link reversal and proteinase K treatment, DNA was extracted with phenol-chloroform, ethanol-precipitated and analyzed by qRT-PCR with specific primers (). Enrichment was calculated as the percentage of input DNA using the formula: % INPUT = 2 exp [Ct (BOUND) – (Ct (UNBOUND) - log2 (UNBOUND DILUTION FACTOR))] × 100.
Statistical analysis
All statistical tests were performed using SPSS version 15.0 (SPSS Inc.). Comparisons between groups were performed with the non-parametric Wilcoxon rank test or Student’s t-test when data were normally distributed. Probabilities less than 0.05 were considered significant.
| Abbreviations: | ||
| 5azaC | = | 5-azacytidine |
| ChIP | = | chromatin immunoprecipitation |
| DAC | = | 5-aza-2'-deoxycytidine |
| DNMT | = | DNA methyltransferase |
| HAT | = | histone acetyltransferase |
| HDAC | = | histone deacetylase |
| NKG2D | = | natural-killer group 2, member D |
| NKG2DL | = | NKG2D ligands |
| VPA | = | valproic acid |
Additional material
Download Zip (344.4 KB)Acknowledgments
This work was supported by Spanish grants from the Red de Investigación Renal (REDinREN) and, FIS PI08/0566 and PI12/02587 from the Instituto Carlos III. ABR was supported by a Severo Ochoa fellowship (FICYT, Consejería de Educación y Ciencia del Principado de Asturias, Spain).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/23115
References
- Glienke J, Sobanov Y, Brostjan C, Steffens C, Nguyen C, Lehrach H, et al. The genomic organization of NKG2C, E, F, and D receptor genes in the human natural killer gene complex. Immunogenetics 1998; 48:163 - 73; http://dx.doi.org/10.1007/s002510050420; PMID: 9683661
- López-Larrea C, Suárez-Alvarez B, López-Soto A, López-Vázquez A, Gonzalez S. The NKG2D receptor: sensing stressed cells. Trends Mol Med 2008; 14:179 - 89; http://dx.doi.org/10.1016/j.molmed.2008.02.004; PMID: 18353724
- Maccalli C, Scaramuzza S, Parmiani G. TNK cells (NKG2D+ CD8+ or CD4+ T lymphocytes) in the control of human tumors. Cancer Immunol Immunother 2009; 58:801 - 8; http://dx.doi.org/10.1007/s00262-008-0635-x; PMID: 19089424
- Fasth AE, Björkström NK, Anthoni M, Malmberg KJ, Malmström V. Activating NK-cell receptors co-stimulate CD4(+)CD28(-) T cells in patients with rheumatoid arthritis. Eur J Immunol 2010; 40:378 - 87; http://dx.doi.org/10.1002/eji.200939399; PMID: 19904767
- Pariente B, Mocan I, Camus M, Dutertre CA, Ettersperger J, Cattan P, et al. Activation of the receptor NKG2D leads to production of Th17 cytokines in CD4+ T cells of patients with Crohn’s disease. Gastroenterology 2011; 141:217 - 26, 226, e1-2; http://dx.doi.org/10.1053/j.gastro.2011.03.061; PMID: 21600899
- Li WX, Pan HF, Hu JL, Wang CZ, Zhang N, Li J, et al. Assay of T- and NK-cell subsets and the expression of NKG2A and NKG2D in patients with new-onset systemic lupus erythematosus. Clin Rheumatol 2010; 29:315 - 23; http://dx.doi.org/10.1007/s10067-009-1322-9; PMID: 20012119
- de Menthon M, Lambert M, Guiard E, Tognarelli S, Bienvenu B, Karras A, et al. Excessive interleukin-15 transpresentation endows NKG2D+CD4+ T cells with innate-like capacity to lyse vascular endothelium in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheum 2011; 63:2116 - 26; http://dx.doi.org/10.1002/art.30355; PMID: 21484763
- Sáez-Borderías A, Gumá M, Angulo A, Bellosillo B, Pende D, López-Botet M. Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur J Immunol 2006; 36:3198 - 206; http://dx.doi.org/10.1002/eji.200636682; PMID: 17109473
- Wang Y, Bai J, Li F, Wang H, Fu X, Zhao T, et al. Characteristics of expanded CD4+CD28null T cells in patients with chronic hepatitis B. Immunol Invest 2009; 38:434 - 46; http://dx.doi.org/10.1080/08820130902943105; PMID: 19811419
- Alonso-Arias R, Moro-García MA, López-Vázquez A, Rodrigo L, Baltar J, García FM, et al. NKG2D expression in CD4+ T lymphocytes as a marker of senescence in the aged immune system. Age (Dordr) 2011; 33:591 - 605; http://dx.doi.org/10.1007/s11357-010-9200-6; PMID: 21210234
- Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL-15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004; 21:357 - 66; http://dx.doi.org/10.1016/j.immuni.2004.06.020; PMID: 15357947
- Muntasell A, Magri G, Pende D, Angulo A, López-Botet M. Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood 2010; 115:5170-9.
- Ventre E, Brinza L, Schicklin S, Mafille J, Coupet CA, Marçais A, et al. Negative regulation of NKG2D expression by IL-4 in memory CD8 T cells. J Immunol 2012; 189:3480 - 9; http://dx.doi.org/10.4049/jimmunol.1102954; PMID: 22942430
- Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002; 419:734 - 8; http://dx.doi.org/10.1038/nature01112; PMID: 12384702
- Peraldi MN, Berrou J, Dulphy N, Seidowsky A, Haas P, Boissel N, et al. Oxidative stress mediates a reduced expression of the activating receptor NKG2D in NK cells from end-stage renal disease patients. J Immunol 2009; 182:1696 - 705; PMID: 19155520
- Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006; 108:4118 - 25; http://dx.doi.org/10.1182/blood-2006-03-006700; PMID: 16902152
- Burgess SJ, Marusina AI, Pathmanathan I, Borrego F, Coligan JE. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J Immunol 2006; 176:1490 - 7; PMID: 16424177
- Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica 2012; 97:1295 - 303; http://dx.doi.org/10.3324/haematol.2011.058529; PMID: 22491735
- González S, López-Soto A, Suarez-Alvarez B, López-Vázquez A, López-Larrea C. NKG2D ligands: key targets of the immune response. Trends Immunol 2008; 29:397 - 403; http://dx.doi.org/10.1016/j.it.2008.04.007; PMID: 18602338
- Eagle RA, Traherne JA, Hair JR, Jafferji I, Trowsdale J. ULBP6/RAET1L is an additional human NKG2D ligand. Eur J Immunol 2009; 39:3207 - 16; http://dx.doi.org/10.1002/eji.200939502; PMID: 19658097
- Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med 2005; 202:583 - 8; http://dx.doi.org/10.1084/jem.20050994; PMID: 16129707
- Zloza A, Lyons GE, Chlewicki LK, Kohlhapp FJ, O’Sullivan JA, Lacek AT, et al. Engagement of NK receptor NKG2D, but not 2B4, results in self-reactive CD8+ T cells and autoimmune vitiligo. Autoimmunity 2011; 44:599 - 606; http://dx.doi.org/10.3109/08916934.2011.593599; PMID: 21913803
- Suárez-Álvarez B, Fernández-Sánchez A, López-Vázquez A, Coto E, Ortega F, López-Larrea C. NKG2D and its ligands: active factors in the outcome of solid organ transplantation?. Kidney Int Suppl 2011; 1:52 - 7; http://dx.doi.org/10.1038/kisup.2011.13
- Kaplan MJ, Lu Q, Wu A, Attwood J, Richardson B. Demethylation of promoter regulatory elements contributes to perforin overexpression in CD4+ lupus T cells. J Immunol 2004; 172:3652 - 61; PMID: 15004168
- Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, et al. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J Immunol 2006; 176:4083 - 93; PMID: 16547244
- Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem 2007; 282:5969 - 72; http://dx.doi.org/10.1074/jbc.C600322200; PMID: 17218320
- Li G, Yu M, Weyand CM, Goronzy JJ. Epigenetic regulation of killer immunoglobulin-like receptor expression in T cells. Blood 2009; 114:3422 - 30; http://dx.doi.org/10.1182/blood-2009-01-200170; PMID: 19628706
- Juelich T, Sutcliffe EL, Denton A, He Y, Doherty PC, Parish CR, et al. Interplay between chromatin remodeling and epigenetic changes during lineage-specific commitment to granzyme B expression. J Immunol 2009; 183:7063 - 72; http://dx.doi.org/10.4049/jimmunol.0901522; PMID: 19915065
- Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol 2010; 22:583 - 91; http://dx.doi.org/10.1016/j.coi.2010.08.013; PMID: 20869864
- Garcia-Manero G, Fenaux P. Hypomethylating agents and other novel strategies in myelodysplastic syndromes. J Clin Oncol 2011; 29:516 - 23; http://dx.doi.org/10.1200/JCO.2010.31.0854; PMID: 21220589
- Kang J, Chen J, Shi Y, Jia J, Zhang Y. Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol 2005; 69:1205 - 13; http://dx.doi.org/10.1016/j.bcp.2005.01.014; PMID: 15794941
- Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285:727 - 9; http://dx.doi.org/10.1126/science.285.5428.727; PMID: 10426993
- Suárez-Alvarez B, López-Vázquez A, Díaz-Molina B, Bernardo-Rodríguez MJ, Alvarez-López R, Pascual D, et al. The predictive value of soluble major histocompatibility complex class I chain-related molecule A (MICA) levels on heart allograft rejection. Transplantation 2006; 82:354 - 61; http://dx.doi.org/10.1097/01.tp.0000228911.22944.23; PMID: 16906033
- Araki Y, Fann M, Wersto R, Weng NP. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: perforin and granzyme B). J Immunol 2008; 180:8102 - 8; PMID: 18523274
- Liu Y, Kuick R, Hanash S, Richardson B. DNA methylation inhibition increases T cell KIR expression through effects on both promoter methylation and transcription factors. Clin Immunol 2009; 130:213 - 24; http://dx.doi.org/10.1016/j.clim.2008.08.009; PMID: 18945643
- Liu Y, Chen Y, Richardson B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4(+)CD28(-) T cells. Clin Immunol 2009; 132:257 - 65; http://dx.doi.org/10.1016/j.clim.2009.03.529; PMID: 19394279
- Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem 2004; 279:51163 - 71; http://dx.doi.org/10.1074/jbc.M409024200; PMID: 15383533
- Gao XN, Lin J, Wang LL, Yu L. Demethylating treatment suppresses natural killer cell cytolytic activity. Mol Immunol 2009; 46:2064 - 70; http://dx.doi.org/10.1016/j.molimm.2009.02.033; PMID: 19394699
- Schmiedel BJ, Arélin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer 2011; 128:2911 - 22; http://dx.doi.org/10.1002/ijc.25635; PMID: 20960460
- Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl J Jr.. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett 2007; 581:1317 - 22; http://dx.doi.org/10.1016/j.febslet.2007.02.045; PMID: 17349632
- Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N. Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J Immunol 2010; 185:5693 - 703; http://dx.doi.org/10.4049/jimmunol.1002188; PMID: 20926794
- Tang F, Chen Z, Ciszewski C, Setty M, Solus J, Tretiakova M, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J Exp Med 2009; 206:707 - 19; http://dx.doi.org/10.1084/jem.20071887; PMID: 19237603
- Ito A, Shimura H, Nitahara A, Tomiyama K, Ito M, Kanekura T, et al. NK cells contribute to the skin graft rejection promoted by CD4+ T cells activated through the indirect allorecognition pathway. Int Immunol 2008; 20:1343 - 9; http://dx.doi.org/10.1093/intimm/dxn092; PMID: 18697765
- Feng L, Ke N, Ye Z, Guo Y, Li S, Li Q, et al. Expression of NKG2D and its ligand in mouse heart allografts may have a role in acute rejection. Transplant Proc 2009; 41:4332 - 9; http://dx.doi.org/10.1016/j.transproceed.2009.08.060; PMID: 20005394
- Joshi M, Dindelegan G, Olausson M, Oltean M. Natural killer group 2 member D cell recruitment driven by major histocompatibility complex class I chain-related antigens A and B: a possible mechanism during acute intestinal allograft rejection in the mouse. Transplant Proc 2010; 42:4467 - 9; http://dx.doi.org/10.1016/j.transproceed.2010.09.115; PMID: 21168719
- Gannagé M, Buzyn A, Bogiatzi SI, Lambert M, Soumelis V, Dal Cortivo L, et al. Induction of NKG2D ligands by gamma radiation and tumor necrosis factor-alpha may participate in the tissue damage during acute graft-versus-host disease. Transplantation 2008; 85:911 - 5; http://dx.doi.org/10.1097/TP.0b013e31816691ef; PMID: 18360276
- Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity 2004; 20:757 - 67; http://dx.doi.org/10.1016/j.immuni.2004.05.008; PMID: 15189740
- Ito Y, Kanai T, Totsuka T, Okamoto R, Tsuchiya K, Nemoto Y, et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am J Physiol Gastrointest Liver Physiol 2008; 294:G199 - 207; http://dx.doi.org/10.1152/ajpgi.00286.2007; PMID: 17962357
- Andersson AK, Sumariwalla PF, McCann FE, Amjadi P, Chang C, McNamee K, et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: a potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum 2011; 63:2617 - 29; http://dx.doi.org/10.1002/art.30460; PMID: 21618462
- Ogasawara K, Benjamin J, Takaki R, Phillips JH, Lanier LL. Function of NKG2D in natural killer cell-mediated rejection of mouse bone marrow grafts. Nat Immunol 2005; 6:938 - 45; http://dx.doi.org/10.1038/ni1236; PMID: 16086018
- Li J, Zhu H, Wang S, Ye P, Liu C, Wu J, et al. Blockade of NKG2D synergized with CTLA4-Ig in promoting long-term graft survival in murine models of cardiac transplantation. Transplantation 2012; 93:356 - 63; http://dx.doi.org/10.1097/TP.0b013e31823ffce7; PMID: 22179404
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132:365 - 86; PMID: 10547847
- Kottilil S, Shin K, Jackson JO, Reitano KN, O’Shea MA, Yang J, et al. Innate immune dysfunction in HIV infection: effect of HIV envelope-NK cell interactions. J Immunol 2006; 176:1107 - 14; PMID: 16393999
- Suárez-Alvarez B, Rodriguez RM, Calvanese V, Blanco-Gelaz MA, Suhr ST, Ortega F, et al. Epigenetic mechanisms regulate MHC and antigen processing molecules in human embryonic and induced pluripotent stem cells. PLoS One 2010; 5:e10192; http://dx.doi.org/10.1371/journal.pone.0010192; PMID: 20419139