Abstract
Glioblastoma (GBM) is the most common and malignant type of primary brain tumor in adults and prognosis of most GBM patients is poor. However, a small percentage of patients show a long term survival of 36 mo or longer after diagnosis. Epigenetic profiles can provide molecular markers for patient prognosis: recently, a G-CIMP positive phenotype associated with IDH1 mutations has been described for GBMs with good prognosis. In the present analysis we performed genome-wide DNA methylation profiling of short-term survivors (STS; overall survival < 1 y) and long-term survivors (LTS; overall survival > 3 y) by utilizing the HumanMethylation450K BeadChips to assess quantitative methylation at > 480,000 CpG sites. Cluster analysis has shown that a subset of LTS showed a G-CIMP positive phenotype that was tightly associated with IDH1 mutation status and was confirmed by analysis of the G-CIMP signature genes. Using high stringency criteria for differential hypermethylation between non-cancer brain and tumor samples, we identified 2,638 hypermethylated CpG loci (890 genes) in STS GBMs, 3,101 hypermethylated CpG loci (1,062 genes) in LTS (wild type IDH1) and 11,293 hypermethylated CpG loci in LTS (mutated for IDH1), reflecting the CIMP positive phenotype. The location of differentially hypermethylated CpG loci with respect to CpG content, neighborhood context and functional genomic distribution was similar in our sample set, with the majority of CpG loci residing in CpG islands and in gene promoters. Our preliminary study also identified a set of CpG loci differentially hypermethylated between STS and LTS cases, including members of the homeobox gene family (HOXD8, HOXD13 and HOXC4), the transcription factors NR2F2 and TFAP2A, and Dickkopf 2, a negative regulator of the wnt/β-catenin signaling pathway.
Introduction
Glioblastoma (GBM) is the most common and most malignant primary brain tumor in adults. Despite recent advances in treatment, including surgical resection, local radiotherapy and systematic chemotherapy, the median survival time after diagnosis is 9–12 mo. However, a small fraction (3–5%) of patients survive for 36 mo or longer. These patients are known as long-term survivors (LTS).Citation1 LTS patients are usually younger in age and have good Karnofsky performance status at the time of diagnosis and, most often, have tumors with promoter hypermethylation of the MGMT gene.Citation2 Recently, The Cancer Genome Atlas (TCGA) research network identified a CpG island methylator phenotype (CIMP) in a subset of gliomas with distinct clinical and molecular features. G-CIMP positive status is associated with mutations in the isocitrate dehydrogenase 1 and 2 genes (IDH1 and IDH2) and linked to a more favorable survival outcome.Citation3 A more recent paper has demonstrated that IDH1 mutation is sufficient to establish the glioma-CIMP phenotype by remodeling the methylome.Citation4 In order to understand more fully the molecular mechanisms underlying the establishment of LTS and STS gliomas we undertook an epigenetic approach. We used the latest Illumina BeadChip technology, which allows interrogation of > 480,000 CpG (HumanMethylation450 BeadChips) sites per sample to determine the methylome of STS and LTS gliomas and to identify differentially methylated CpG loci.
Results and Discussion
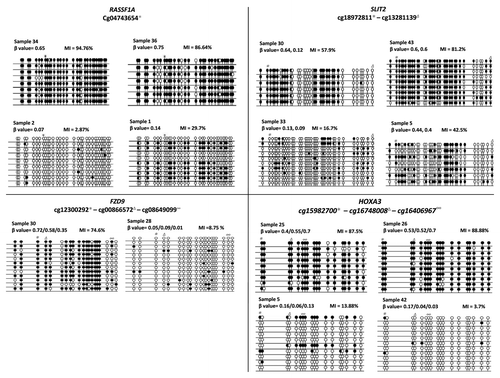
HumanMethylation450 BeadChips were used to analyze methylation profiles of 19 STS and 16 LTS grade IV glioblastomas as well as DNA samples from normal human brain (n = 4). Initial analysis involved unbiased validation of the array data by cloning and bisulfite sequencing of previously identified frequently hypermethylated genes in gliomas (RASSF1A, SLIT2, HOXA3 and FZD9).Citation5-Citation7 demonstrates that high β-values correspond to a high methylation index while low β-values correspond to a low methylation index (). Since there are now multiple studies validating the 450k Illumina array, we felt comfortable using this data for STS/LTS glioma methylation profiling.Citation8-Citation10
Figure 1. Validation of probe β value using bisulfite clone sequencing. Individual samples were tested with high or with low β values for each of the 4 genes, HOXA3, SLIT2, FZD9 and RASSF1A. For each sample and loci, the β value is given and the methylation index (MI) was calculated using the formula, number of CpG dinucleotides methylated/total number of CpG dinucleotides sequenced × 100. Black circles represent methylation of the CpG loci and white circles represent unmethylated loci.
CIMP in a subset of LTS cases
Unsupervised hierarchical clustering of the 500 most variable loci indicated a CIMP positive (CIMP+) phenotype in five LTS tumors (). IDH1 and IDH2 mutation analysis revealed that the 5 LTS with CIMP+ phenotype all had IDH1 mutation, no IDH1 or IDH2 mutations were found in the rest of the tumor cohort. Hence, CIMP+ phenotype is tightly associated with IDH mutations, which agrees with other recent studies.Citation3,Citation4 We further confirmed the CIMP+ phenotype by looking at methylation of the G-CIMP+ signature genes identified in the Noushmehr et al., 2010Citation3 study (Fig. S1). Although residing in the same large cluster as the LTS CIMP-ve samples, the majority of STS tumors clustered next to each other. Clustering of the non-CIMP tumors demonstrated more subtle differences between STS and non-CIMP LTS samples (Fig. S2).
Figure 2. Unsupervised clustering of the 500 most variable CpG loci identifies CIMP+ phenotype in 5 LTS tumors. CpG loci are color coded from white (low methylation) to black (hypermethylation). The bar below the figure indicates CIMP status (black, CIMP+; white, CIMP-), IDH mutation (black, mutated; white, wild-type), MGMT methylation (black, methylated; white, unmethylated; crossed, undetermined), and type of glioma (white, STS; black, LTS).

Methylome of STS gliomas
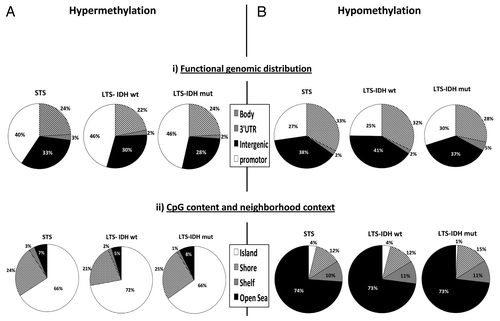
Following removal of all probes showing β-value > 0.25 in any of the four normal samples, the remaining probes were considered hypermethylated if > 30% of tumor samples had a β-value of > 0.5. Following this criterion, 2,638 CpG loci (corresponding to 890 genes) were identified as hypermethylated in the STS cases. Functional genomic distribution identified that 40% (1,064) of the hypermethylated CpG loci resided in gene promoter regions (CpG sites located within 200 bp or 1,500 bp of the transcription start site (TSS) and in the 5′ untranslated region and exon 1). From the CpG content and neighborhood context majority, 66% (1,747) of CpG loci resided in CpG islands (). We were able to confirm the top-80 hypermethylated CpG loci in the TCGA data set (Fig. S3). Due to the extensive coverage of CpG loci in the 450k array, we were able to further refine our gene list to contain only genes exhibiting frequent hypermethylation in multiple probes, demonstrating a wider level of hypermethylation throughout the region. Removal of genes represented by < 3 probes resulted in 272 hypermethylated probes corresponding to 49 genes (Table S1). DAVID functional annotation identified enrichment for genes involved in regulation of cell development, cell migration, cell-cell adhesion and transcription factors.
Figure 3. Description of the methylome of the 3 groups of glioma in the (A) hypermethylated, (B) hypomethylated CpG loci. Functional genomic distribution: body, 3′UTR, intergenic and promoter region (i). CpG content and neighborhood context classification: island, shore, shelf and open sea (ii).
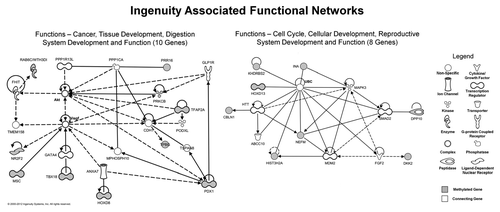
Ingenuity analysis highlighted the majority of STS hypermethylated genes as being associated with gene expression and/or cellular development (). Genes also fell into functional networks, the top two of which are involved with, among others, cancer, tissue development and cell cycle and cellular development (Fig. S4).
Table 1. Ingenuity analysis of the STS genes demonstrating the associated cancer genes and top 5 molecular and cellular functions
We also identified a set of 946 hypomethylated CpG loci in STS cases. The majority of the hypomethylated loci resided in the body of the genes (33%) and in the intergenic regions (38%). In relation to the CpG content and neighborhood context, 74% of the hypomethylated loci were located in Open Seas, and only 4% resided in CpG islands ().
Methylome of LTS gliomas with and without IDH1 mutation
The 5 CIMP+ LTS cases, all with mutated IDH1 gene, showed a much higher number of hypermethylated CpG loci [11,293 (in 3,323 genes)], compared with STS or LTS with wild type IDH1 gene (2,638 and 3,101, respectively). The location of hypermethylated loci was similar between STS and LTS (with or without IDH mutation) samples. In order to identify CIMP+ genes, we looked for differentially hypermethylated probes between LTS with IDH mutated genes and the remaining IDH wild type samples. Using FDR adjusted p value of < 0.05 between IDH mutated and IDH wild type gliomas and genes represented by > 3 CpG probes, we identified 535 genes that represent CIMP+ phenotype (Table S2). DAVID functional analysis indicated these genes to be involved in cell-cell signaling, cell adhesion, regulation of nervous system development and apoptosis. Ingenuity analysis identified 31% (166/535) of these genes to be associated with cancer, a similar percentage to that seen in STS hypermethylated loci (Tables S3, S4 and S5). The top five molecular and cellular functions of the CIMP genes encompassed between 19.7% and 31% of genes in each of these categories and consisted of genes involved in cellular movement, cellular growth and proliferation, cell morphology, cellular assembly and organization, and cellular function and maintenance. Fifty genes were present in all five categories (Table S3). The top functional network contained 5.6% (30/535) of genes and is involved in cell morphology, cell death and survival and renal necrosis/cell death (Fig. S5). Additionally, due to the large number of CIMP genes, we were able to look for pathways showing hypermethylation of multiple members and identified glutamate receptor signaling as having multiple methylated targets (Fig. S6). In total, 1,402 hypomethylated loci were identified. The majority of them resided in the intergenic regions (37%), 73% were located in Open Seas and only 1% was found CpG islands.
Due to the lack of CIMP+, in LTS cases without IDH1/2 mutations we saw a more similar level of hypermethylated loci as in STS samples. In total, we observed 3,101 hypermethylated CpG loci corresponding to 1,062 genes: 46% of these were located in gene promoters and 72% were located in CpG islands. We also observed 690 hypomethylated CpG loci, which, again, showed a similar distribution to STS and LTS IDH mutated samples ().
Identification of hypermethylated loci in STS vs. LTS
In order to identify epigenetic markers for poor prognosis in STS cases that are likely to be biologically relevant, we looked for differential hypermethylation in STS vs. LTS cases among CpG probes located in gene promoters (TSS1500, TSS200, 5′UTR and 1st exon). Using a FDR corrected p value cut off < 0.05 and at least a 0.2 β-value difference between median β-values for STS vs. LTS samples, we identified 32 CpG loci (representing 23 genes) that exhibited significantly increased methylation in STS cases (). Ingenuity analysis identified 43.5% (10/23) genes to have an association with cancer (). Some of these genes have previously been shown to be hypermethylated in brain and other cancers (e.g., HOX genes and TFAP2A), but other genes, to the best of our knowledge, have not been previously associated with methylation in gliomas and/or other cancers (e.g., CBLN1 and CBLN4, ISM1). Univariate survival analysis confirmed poor prognosis for NR2F2 and the other top hypermethylated loci, INA (Fig. S7). We did not find any striking differences between the molecular and cellular functions of the top genes within the list of STS vs. LTS methylated genes compared with the list of STS and LTS hypermethylated genes when analyzed separately. The top two functional networks included cancer, tissue development, cell cycle and cellular development ().
Table 2. List of the most significantly and differentially hypermethylated genes in STS vs. LTS with FDR adjusted P value of < 0.05
Table 3. Ingenuity analysis of the STS vs. LTS differentially methylated genes demonstrating the associated cancer genes and top molecular and cellular functions
Figure 4. Top two gene networks generated from Ingenuity analysis of the STS vs. LTS differentially hypermethylated genes. Of the 23 STS differentially hypermethylated genes, 18 genes fell within these two networks. Methylated genes are shaded gray, while the necessary connecting genes are unshaded.
In order to assess the biological relevance of gene methylation, we analyzed the expression of a subset of STS vs. LTS differentially methylated genes in a panel of glioma cell lines treated with 5-aza-2-deoxycytidine. Expression of DKK2, TFAP2A and NR2F2 was upregulated in methylated glioma cell lines post 5-aza-dC treatment, hence confirming the functional significance of DNA methylation in these genes (Fig. S8).
In summary, we have established: (1) the methylomes of poor and good prognosis glioblastomas using the latest high density methylation BeadChips; (2) a tight association of CIMP+ with IDH1 mutations in a subset of good prognosis gliomas—and have identified additional CIMP+ genes; (3) a preliminary set of CpG loci that show differential hypermethylation in STS compared with LTS gliomas—methylation of some of these loci may be biologically relevant. Future studies will be required using much larger sample cohorts with comparable clinical parameters to determine the relevance of these differentially methylated loci.
Materials and Methods
DNA samples
DNA from 35 grade IV glioblastoma patients were used in this study (Table S6). These samples include 19 short-term survival (STS) glioblastomas (overall survival < 1year) and 16 long-term survival (LTS) glioblastomas (overall survival > 3 y). DNA from 4 non-cancerous normal brain samples were also included. All samples were collected with ethical approval following institutional guidelines and informed patient consent.
Illumina Infinium HumanMethylation450K BeadChips (Illumina)
Bisulfite modification of DNA and array hybridization was performed by Cambridge Genomics Services. Normalized data was obtained using Genome Studio software from Illumina. A pipeline was designed using the methods available in the R(bioconductor) package lumi, to correct the color bias using a quantile approach.Citation11 The two types of probes (Infinium Type I and II) were separated and then both color channel were normalized using a quantile approach, this removes the color bias by giving the two channels the same distribution within the same array. Then, the data from the type I and II probes were merged together to recreate the set and between array normalization was applied using a quantile approach. Finally, the β score were calculated for each probe using the formula provided by Illumina [if M is the methylated intensity and U the umethylated intensity: Beta = M/(U + M + 100)].
Data processing
Probes demonstrating detection p values greater than 0.01 in any sample were removed along with probes located on the X and Y chromosomes, probes with a single nucleotide polymorphism (SNP) and probes for imprinted genes were also removed (remaining probes = 440,287). We analyzed the data for hypermethylation and hypomethylation separately. To ensure tumor specific hypermethylation, probes showing a β-value > 0.25 in any of the four normal samples were also removed. Hypermethylation was subsequently determined as a β-value > 0.5. This was considered relevant if present in > 30% tumor samples. After applying these filtrations, 133,975 CpG loci were used subsequently in this study. On the other hand, Hypomethylation was considered significant if β-value < 0.25 and present in > 30% of tumor samples and after removing probes with β-value < 0.5 in any of the four normal samples (remaining CpG loci = 183,737).
G-CIMP
The top 500 most variable loci were determined from the standard deviation. Clustering was performed using a hierarchical Euclidean based algorithm. Further CIMP characterization was determined using probes for G-CIMP genes described by Noushmehr et al., 2010Citation3 (ANKRD43, DOCK5, LGALS3, FAS, HFE, MAL and RHOF).
Methylation and mutation analysis
Clone bisulfite sequencing was used for array validation following the procedure previously discribed.Citation12 MGMT methylation status was determined using methylation specific PCR (MSP) with primers previously described.Citation13 COBRA analysis was performed as described previously.Citation12 Table S7 contains methylation primers used in this study. For IDH1 and IDH2 mutation analysis, previously described primers were used to amplify relevant fragments of the IDH1 and IDH2 genes.Citation14 PCR reactions were performed as described previously.
Cell line expression analyses
Treatment of glioma cell lines with 5′-aza-2′-deoxycitidine (5-aza-dC) and expression analysis were performed as described previously.Citation15
TCGA samples
Illumina Infinium HumanMethylation 450k array data from TCGA website (http://cancergenome.nih.gov) was used for the following 18 TCGA (level 3) primary glioblastomas: (Batch 79: TCGA-76-4928, TCGA-76-4926, TCGA-26-5135, TCGA-12-5301, TCGA-12-5299, TCGA-06-5418; Batch 111: TCGA-32-1980, TCGA-81-5910, TCGA-19-5955, TCGA-19-5951, TCGA-19-5947, TCGA-14-0781, TCGA-06-6391, TCGA-06-5856, TCGA-06-5412, TCGA-06-5411, TCGA-06-5410 and TCGA-06-5408). Cases were selected if survival was less than one year from initial date of diagnosis.
Statistical analysis
Two-tailed student t-test was used to identify differentially hypermethylated CpG loci between CIMP+ and CIMP- groups. For identification of CIMP+ genes, significant changes were considered when p < 0.05 after applying the Benjamini and Hochberg method for False Discovery rate (FDR) correction (Adj.Pval). Differentially hypermethylated CpG loci in STS vs LTS cases were identified using Limma package in R and probes with FDR adjusted p value < 0.05 were considered significant. Hierarchical clustering was performed using Euclidean distance for the most 500 variables CpG loci defined by Standard Deviation (SD). Kaplan-Meier survival curves were produced using SPSS v.18 to determine the association between DNA methylation and patient survival, p < 0.05 was considered significant. Ingenuity pathway analysis (IPA) (www.ingenuity.com) and DAVID analysis (http://david.abcc.ncifcrf.gov/) software were used for gene ontology and pathway analysis for differentially methylated genes.
Additional material
Download Zip (5.7 MB)Acknowledgments
T.S. was sponsored by King Abdulaziz University, Jeddah, Saudi Arabia. V.K.H. was sponsored by the Department of Neurosurgery, University Hospital Dresden, Germany.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al, German Glioma Network. Long-term survival with glioblastoma multiforme. Brain 2007; 130:2596 - 606; http://dx.doi.org/10.1093/brain/awm204; PMID: 17785346
- Hesson LB, Krex D, Latif F. Epigenetic markers in human gliomas: prospects for therapeutic intervention. Expert Rev Neurother 2008; 8:1475 - 96; http://dx.doi.org/10.1586/14737175.8.10.1475; PMID: 18928342
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al, Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17:510 - 22; http://dx.doi.org/10.1016/j.ccr.2010.03.017; PMID: 20399149
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012; 483:479 - 83; http://dx.doi.org/10.1038/nature10866; PMID: 22343889
- Dallol A, Krex D, Hesson L, Eng C, Maher ER, Latif F. Frequent epigenetic inactivation of the SLIT2 gene in gliomas. Oncogene 2003; 22:4611 - 6; http://dx.doi.org/10.1038/sj.onc.1206687; PMID: 12881718
- Hesson L, Bièche I, Krex D, Criniere E, Hoang-Xuan K, Maher ER, et al. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene 2004; 23:2408 - 19; http://dx.doi.org/10.1038/sj.onc.1207407; PMID: 14743209
- Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, et al. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 2009; 4:255 - 64; PMID: 19550145
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692 - 702; http://dx.doi.org/10.4161/epi.6.6.16196; PMID: 21593595
- Cahill N, Bergh AC, Kanduri M, Göransson-Kultima H, Mansouri L, Isaksson A, et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia 2012; In press http://dx.doi.org/10.1038/leu.2012.245; PMID: 22922567
- Roessler J, Ammerpohl O, Gutwein J, Hasemeier B, Anwar SL, Kreipe H, et al. Quantitative cross-validation and content analysis of the 450k DNA methylation array from Illumina, Inc. BMC Res Notes 2012; 5:210; http://dx.doi.org/10.1186/1756-0500-5-210; PMID: 22546179
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24:1547 - 8; http://dx.doi.org/10.1093/bioinformatics/btn224; PMID: 18467348
- Hill VK, Hesson LB, Dansranjavin T, Dallol A, Bieche I, Vacher S, et al. Identification of 5 novel genes methylated in breast and other epithelial cancers. Mol Cancer 2010; 9:51; http://dx.doi.org/10.1186/1476-4598-9-51; PMID: 20205715
- Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350 - 4; http://dx.doi.org/10.1056/NEJM200011093431901; PMID: 11070098
- Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 2009; 118:469 - 74; http://dx.doi.org/10.1007/s00401-009-0561-9; PMID: 19554337
- Hill VK, Underhill-Day N, Krex D, Robel K, Sangan CB, Summersgill HR, et al. Epigenetic inactivation of the RASSF10 candidate tumor suppressor gene is a frequent and an early event in gliomagenesis. Oncogene 2011; 30:978 - 89; http://dx.doi.org/10.1038/onc.2010.471; PMID: 20956940