Abstract
The histone H2A variant H2AZ is an essential chromatin signaling factor. Herein, we report that H2AZ is monomethylated at lysine 7 (H2AZK7me1) by the lysine methyltransferase SETD6. We observed that methylation of H2AZ increased noticeably upon cellular differentiation of mouse embryonic stem cells (mESCs). H2AZK7me1 and the repressive H3K27me3 mark were found near the transcriptional start sites of differentiation marker genes, but were removed upon retinoic acid-induced cellular differentiation. The depletion of Setd6 in mESCs led to cellular differentiation, compromised self-renewal, and poor clonogenicity. These findings demonstrate that mESCs require Setd6 for self-renewal and portray H2AZK7me1 as a marker of cellular differentiation.
Introduction
The role of lysine methylation in human pathologies is highlighted by the increasing number of lysine methyltransferases (KMT) involved in the etiology of cancer,Citation1,Citation2 growth defectsCitation3,Citation4 and neurological disorders.Citation5 Therefore, the identification and characterization of novel KMT substrates is postulated to be pivotal for the understanding of human pathologies.
Our current knowledge of how H2AZ functions to regulate DNA-dependent cellular processes is limited. H2AZ is found at actively transcribed promoters in human cells,Citation6 suggesting a role in favoring open chromatin conformations. Recently, a genome-wide study conducted in human cells by Valdés-Mora et al. showed that the acetylated form of H2AZ was solely found at the transcriptional start site of actively expressed genes, while the deacetylated form of H2AZ localized at silenced promoters.Citation7 Therefore, posttranslational modifications appear to be a critical component of gene expression regulation by H2AZ.
Embryonic stem cells (ESCs) have the potential to differentiate and give rise to all the cell types constituting multicellular organisms, an aptitude known as pluripotency. ESCs are also characterized by their ability to self-renew. It has been shown that murine ESCs (mESCs) cellular differentiation is prevented by silencing the expression of H2AZ.Citation8 In addition, H2AZ co-localizes with the transcriptional silencing H3K27me3 mark and the PRC2 complex subunits SUZ12 and EED in mESC near the transcription start site of developmental genes.Citation8,Citation9 However, although H2AZ is important for PRC2 chromatin association,Citation8 PRC2 subunits are dispensable for H2AZ nucleosome incorporation.Citation10
Herein, we demonstrate that SETD6 monomethylates H2AZ on lysine 7 (H2AZK7me1) in vitro and that SETD6 contributes to H2AZ methylation in vivo. Mass spectrometric analysis detected lysine methylation of H2AZ on both lysine 4 and lysine 7 on in vitro methylated H2AZ as well as on endogenous H2AZ. A substantial increase of H2AZ lysine methylation upon mESC differentiation led us to investigate the potential role of Setd6 in stem cell biology. Depletion of Setd6 in mESC resulted in cellular differentiation, compromised self-renewal, and reduced clonogenicity. Finally, we observed that H2AZK7me1 was evicted along with the silencing mark H3K27me3 from the promoter of differentiation markers upon retinoic acid-induced cellular differentiation.
Results
Identification of H2AZ lysine methyltransferases
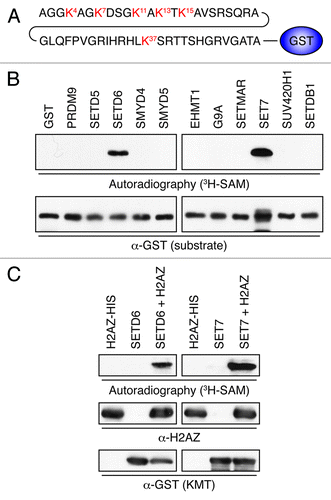
We hypothesized that H2AZ could be modified and regulated by lysine methylation. We thus fused the first 50 residues of H2AZ to the N-terminus of GST (H2AZWT50-GST; ), which was then affinity purified and used as a substrate in an in vitro screen using a panel of KMTs, described previously.Citation11 Each recombinant enzyme was incubated with H2AZWT50-GST in the presence of the tritiated form of the methyl donor S-adenosylmethionine (3H-SAM). H2AZ was methylated predominantly by SETD6 and SET7 (). These were then validated individually on purified full-length H2AZ containing a C-terminal 6xHis tag and we confirmed that both SETD6 and SET7 methylated H2AZ (). These findings suggest that H2AZ is a bona fide substrate of SETD6 and SET7.
Figure 1. Identification of H2AZ lysine methyltransferases. (A) Representation of H2AZWT50-GST. The first 50 amino acids of H2AZ were fused to the N-terminus of GST. (B) Autoradiogram of KMT assays using 3H-SAM, H2AZWT50-GST as a substrate, and the indicated KMTs. The enzymatic reactions were resolved by SDS-PAGE, transferred to PVDF, and either exposed on film (top) or probed with α-GST to show even H2AZ loading. (C) Validation of the potential H2AZ methyltransferases SETD6 and SET7. As in panel B, but full-length recombinant H2AZ-6xHis was used instead of H2AZWT50-GST as the substrate.
H2AZ is methylated on lysines K4 and K7 in vitro
To identify the precise lysine methylation site(s) on H2AZ, all N-terminal lysines were converted to arginines (R) to obtain an unmethylable H2AZ form (K0). Then, each R was individually converted back to K, so that H2AZ contained only one K that could be methylated (). These mutant forms of H2AZ were then subjected to KMT assays with either SETD6 or SET7. The K7K mutant was methylated by SETD6 to the same extent as the wild-type form of H2AZ (WT50) (), suggesting that the SETD6-targeted methylation site is lysine 7. In a similar experiment, K13K appeared to be the major site for SET7 ().
Figure 2. H2AZ is methylated on lysines K4 and K7 in vitro. (A) Representation of the H2AZ mutants. Lysines are highlighted in red. (B) Autoradiogram of the KMT assays using SETD6 (top) or SET7 (bottom) and H2AZ mutants shown in panel A. (C) Flashplate KMT assays using SETD6 and indicated H2AZ-biotin peptides as substrates. Each condition was performed in quadruplicate and the error bars represent the standard error of the mean. The KMT activity is expressed as 3H counts per minute. (D) KMT assays on H2AZWT50-GST were analyzed by immunoblotting using the indicated antibodies. (E) Table summarizing the quantitative MS analysis of SETD6-methylated H2AZ-biotin peptide. (F) Chromatogram of the quantitative MS analysis performed to generate the table in panel E. The molecular weights of mono-methylated K4 and K7 peptides are highlighted (440.29 and 766.49, respectively).
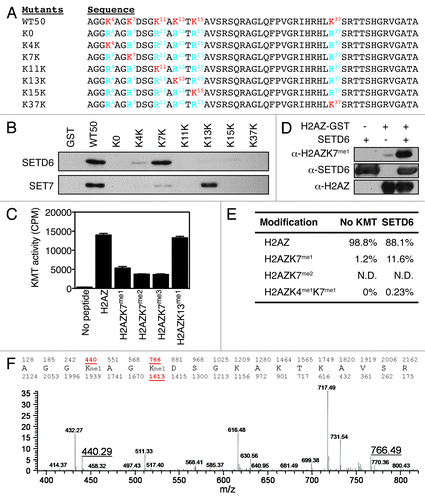
Subsequently, C-terminally biotinylated and mono-, di-, or tri-methylated H2AZK7 peptides were synthesized and subjected to in vitro methylation assays. These assays further demonstrate that H2AZ was effectively methylated by SETD6 (). In agreement with the results in , methylation of H2AZ by SETD6 was hindered by pre-methylation of K7, further suggesting that SETD6 methylates K7 of H2AZ. Specifically, the monomethyl group on K7 impaired the methylation of the peptide by approximately 60%, while the dimethyl modestly impacted H2AZ methylation by an additional 10% decrease in activity, and the trimethyl had no further effect on SETD6 activity (). These results suggest that SETD6 is capable of monomethylating H2AZ. The residual KMT activity on the K7me2 and K7me3 peptides [about 10 times above the no peptide (-) control background in ] suggests that SETD6 may methylate secondary sites such as K4, as observed by a faint methylation signal on the recombinant K4K H2AZ-GST protein (). Equivalent amount of biotinylated peptides were used in the KMT assays (Fig. S1A). In agreement with K7 being the major target site of SETD6, single mutation of K7 to an arginine led to a significant decrease in H2AZ methylation (Fig. S1B). A similar experiment was also conducted with SET7 and the panel of K-R mutants. The latters were all methylated similarly to the wild-type H2AZ substrate (Fig. S1B), suggesting that SET7 indiscriminately methylates multiple sites on H2AZ. Henceforth, we focused our efforts on H2AZK7me1 and SETD6, as little is known about posttranslational modifications of H2AZ, especially lysine methylation, and SETD6 has no reported enzymatic activity on histones.
Recombinant H2AZ was then in vitro methylated by SETD6 and analyzed by immunoblotting using our methyl-specific α-H2AZK7me1 antibody (Fig. S2). The signal from the H2AZK7me1 antibody was substantially increased in the presence of H2AZ and SETD6 (), convincingly indicating that SETD6 monomethylates H2AZ.
To further confirm the site and state of methylation on H2AZ by SETD6, we conducted mass spectrometric (MS) analyses of KMT assays on biotinylated H2AZ peptides. These analyses showed that in vitro SETD6 catalyzes the monomethylation of K7 (H2AZK7me1) while di-methylation on K7 (H2AZK7me2) could not be detected on H2AZ peptides (). Also, low levels of H2AZ methylation were detected simultaneously on both K4 and K7. A detailed MS2 chromatogram highlights the monomethylation of H2AZ at both K4 (molecular mass 440.29) and K7 (molecular mass 766.49) (; Fig. S3). Importantly, monomethylation on K7 was detected on two different peptides (molecular masses 766.49 and 1613.85 in Fig. S3). Altogether, these results show that SETD6 preferentially monomethylates H2AZK7 in vitro, but also methylates secondary sites, such as K4, as detected by mass spectrometric analyses.
H2AZ is methylated on lysines K4 and K7 in vivo
To confirm the biological existence of methyl-H2AZ, in vivo methylation of H2AZ was assessed by immunoblotting total protein lysates from control cells (LVTHM; parental shRNA control vector) and H2AZ depleted cells (shH2AZ). The unmodified-H2AZ and H2AZK7me1 antibodies detected a decrease of H2AZ in the knockdown cell extract (). Importantly, cells depleted of SETD6 by two independent shRNAs (shSETD6 #7 and shSETD6 #10) had reduced levels of H2AZK7me1 (), suggesting that SETD6 monomethylates H2AZ on lysine 7 in vivo.
Figure 3. H2AZ is methylated on lysines K4 and K7 in vivo. (A) Protein extracts from U2OS control (LVTHM) or H2AZ knockdown (shH2AZ) cells were analyzed using the indicated methyl-specific antibody. (B) Lentiviruses were used to transduce U2OS cells to express the indicated human SETD6-targeting shRNAs and protein extracts were analyzed by immunoblotting using the indicated antibodies. (C) Immunoblot analysis of protein extracts from shNanog mESC shows loss of Nanog throughout the differentiation time course. (D) Quantitative MS analyses of the H2AZ peptides derived from differentiating mESC induced by Nanog knockdown at the indicated times. *p value of 0.05. (E) Immunoblot analysis of H2AZK7me1 levels in untreated and RA-treated E14 cells. (F) Quantification by MS of total H2AZ, H2AZK7me1, and H2AZK4me1K7me1 from E14 mESC differentiated with RA. * and ** denote p values of < 0.05 and < 0.02, respectively. (G) Quantification by MS of H2AZ acetylation in mESC during shNanog-induced differentiation shown as Abundance (left) and Fold increase (right).
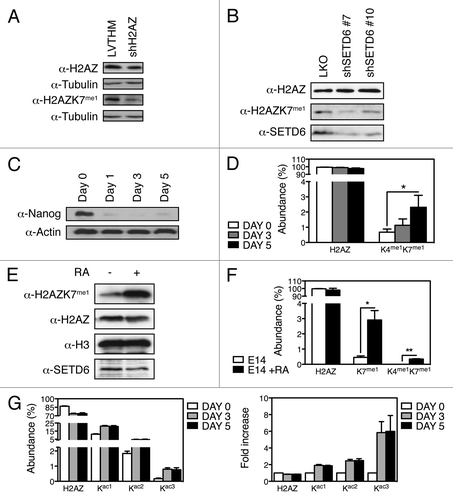
To detect dynamic changes in H2AZ lysine methylation in an in vivo biological system that requires H2AZ, we used an shRNA-mediated methodCitation12 (Fig. S4A) for the depletion of Nanog () and induction of mESC differentiation (Figs. S4B and C). Subsequently, histones from mESCs were analyzed by MS to validate the existence of methyl-H2AZ in vivo. Interestingly, H2AZK7me1 was detected solely in conjunction with H2AZK4me1 (). But, most importantly, methylation of H2AZ at K4 and K7 was increased by over 3-fold following mESC differentiation (; Fig. S4D). As observed with the shNanog-induced cellular differentiation system, induction of cellular differentiation by retinoic acid (RA) led to a noticeable increase in H2AZK7me1 and in H2AZK4me1K7me1 detected by immunoblotting of H2AZK7me1 () and by mass spectrometry (), while the global levels of H2AZ and SETD6 appeared to diminish (). Together, these data demonstrate that H2AZ is monomethylated in vivo on lysines K4 and K7. To conclude, the levels of H2AZK4me1K7me1 increased noticeably in response to cellular differentiation induced by either RA or the silencing of Nanog expression.
Since H2AZ is acetylated on lysines K4, K7, K11, K13 and K15,Citation7,Citation13 we also investigated lysine acetylation during mESC differentiation. Interestingly, we observed that H2AZ acetylation at lysines K4, K7, and K11 increased substantially following Nanog depletion. Specifically, the mono- and di-acetylated forms increased by about 2-fold, from roughly 10% to 20% abundance and from approximately 2% to 4% abundance, respectively. However, the triacetylated (H2AZK4acK7acK11ac) form increased close to 6-fold from 0.15% to 0.8% abundance (). Since the MS analysis did not detect the presence of lysine acetylation and methylation simultaneously on the same peptide, our data indicate that the two modifications on H2AZ are mutually exclusive.
SETD6 is required to maintain self-renewal of mESCs
Because of the compelling data showing an increase of H2AZK4me1K7me1 upon mESC differentiation, we further investigated the potential role of Setd6 and methyl-H2AZ in biological mESC properties. E14 mESC were transduced with shRNA-expressing lentiviral particles to target Setd6. While control cells retained mESC morphology, Setd6-depleted cells appeared differentiated (). Specifically, Setd6 depletion induced a significant increase (p value 0.008) in partially and fully differentiated colonies accompanied by a drastic decrease (p value 0.01) in the undifferentiated colonies (). Setd6 depletion was validated by immunoblotting () and differentiation of shSetd6 mESC confirmed by decreased Nanog level ().
Figure 4. Setd6 maintains mESC self-renewal. (A) E14 mESC were transduced with either shLuc- or shSetd6-expressing lentiviral particles, selected with puromycin, and stained with alkaline phosphatase (AP). AP-stained colonies were scored as fully differentiated (FD), partially differentiated (PD), or undifferentiated (UD) according to the legend. *Denotes a p value of 0.008. (B) Setd6, Nanog, and Tubulin immunoblots from control and shSetd6 mESC. (C) Competition assay in which mCherry-expressing cells (shLuc or shSetd6) were mixed with control cells at a 4:1 ratio, maintained over the course of 6 passages, and monitored for mCherry expression by FACS. *Denotes a p value of < 0.004. (D) Clonogenicity assays in which shLuc, shSetdb1, and shSetd6 cells were plated at clonal density and stained for AP after 10 d. *Denotes a p value of < 0.0002. (E) qPCR validation of Setd6 knockdown and analysis of Fgf5 expression. (F) ChIP analysis of chromatin approximately 500 base pairs upstream of Fgf5 transcriptional start site (TSS) in undifferentiated mESC (E14) or retinoic acid-induced differentiated mESC (E14 + RA). The error bars represent the standard error of the mean calculated from a technical replicate triplicate from a representative biological replicate. *Denotes a p value of < 0.02 between the IgG signal and H2AZK7me1 signal. **Denotes a p value of < 0.05 between the E14 untreated control and the E14 + RA samples.
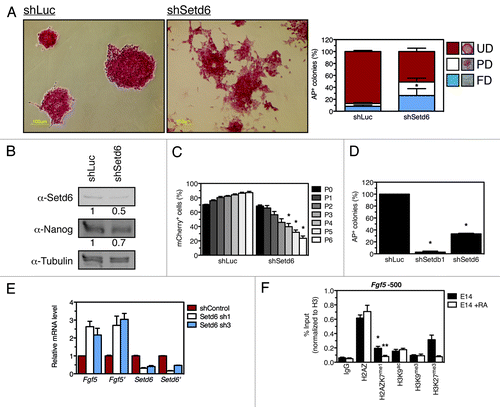
To further investigate the role of Setd6 expression in mESC biology, we conducted a competition assay, as described previously,Citation12,Citation14,Citation15 in which mCherry-expressing (mCherry+) shLuc control or Setd6-depleted mCherry+ populations were each mixed with non-fluorescent mESC (Fig. S5) and maintained over the course of 6 passages. The mixed populations were analyzed for mCherry signal by fluorescence-activated cell sorting (FACS) at each passage. This experiment demonstrates that self-renewal is compromised in the Setd6-depleted population (). Specifically, at passage 0 the population was composed of about 70% Setd6-depleted cells, but these contributed to less than 25% of the population by passage 6 (; Fig. S5). Clonogenicity assays in which depletion of Setdb1 was used as a positive control,Citation16 confirmed that depletion of Setd6 led to impaired mESC self-renewal (). To refute the possibility that the impaired self-renewal of Setd6-depleted cells could be due to cell death, shSetd6 cells were stained for Annexin V (Fig. S6). Also, the Setd6-depleted population had a normal cell cycle profile, which was measured by FACS (Fig. S7), and unaltered proliferation capacity, which was assessed by Ki67 staining (Fig. S8). These results reiterateCitation16 and validate that Setd6 is required for mESC self-renewal.
In response to Setd6 depletion, the expression of differentiation and pluripotency markers was analyzed and we found that Fgf5 was consistently and reproducibly upregulated (). The efficiency of Setd6 knockdown was also confirmed by qPCR (). These results suggest that Setd6 functions to maintain mESC self-renewal, presumably by repressing the expression of differentiation genes.
To address the possibility that H2AZ and H2AZK7me1 could be involved in the regulation of Fgf5 expression, we induced E14 differentiation using retinoic acid (RA). We then investigated the incorporation of H2AZ forms at chromatin at the Fgf5 locus and found by chromatin immunoprecipitation assays that H2AZ was integrated within nucleosomes about 500 base pairs upstream of the Fgf5 transcriptional start site (TSS; ) as well as at the TSS itself (Fig. S9). However, H2AZK7me1 was enriched by about 3-fold at the -500 site and essentially absent from the TSS (; Fig. S9). Interestingly, upon RA-mediated differentiation, the levels of H2AZK7me1 dropped substantially in correlation with the loss of the epigenetic silencing mark H3K27me3 (). In addition, H2AZK7me1 was enriched at the TSS of FoxA2 (4-fold) and Hand2 (3-fold), but depleted along with H3K27me3 upon RA-mediated differentiation (Fig. S9). The RA treatment also induced increased acetylation of the active transcription H3K9ac mark at the TSS of FoxA2 and Hand2 (Fig. S9).
Discussion
Herein, we have identified two potent lysine methyltransferases, SETD6 and SET7, that modify the histone variant H2AZ. Although H2AZ is known to be polyacetylated within the N-terminal tail (at residues K4, K7, K11, K13 and K15)Citation7,Citation13 as well as monoubiquitylated at the carboxy-terminus (at residues K120 and K121),Citation17,Citation18H2AZ had never been reported to be methylated. Interestingly, our methyl-specific H2AZK7me1 antibody detects a slow-migrating band (not shown), which, based on the observed molecular weight, is likely an ubiquitylated form of H2AZ. These observations suggest that lysine methylation and ubiquitylation may occur together and that there could be some form of cross-talk between the two modifications.
Regarding H2AZ acetylation, we anticipate that methylation of H2AZ at lysines 4 and 7 could prevent subsequent acetylation at these sites, thereby impairing the reported transcriptional functions associated with the acetylation of H2AZ.Citation7 Indeed, mass spectrometry analysis did not detect any acetylation on the endogenous H2AZ.
Mass spectrometry data from shNanog and RA-treated cells, in addition to the data using our methyl-specific H2AZK7me1 antibody, highlight the authentic existence of a novel posttranslational modification on H2AZ in human and murine cells. Interestingly, upon RA-induced differentiation, H2AZK7me1 levels increased drastically although SETD6 decreased slightly at the protein level () and mRNA level (not shown). It will be interesting to investigate if SETD6 enzymatic activity is increased upon RA treatment or whether other KMTs are induced by RA. Indeed, we observed increased expression of several KMT in response to RA (not shown). Notably, SET7 mRNA levels increased by about 2-fold in response to RA (not shown) and, as a H2AZ methyltransferase, may thus contribute to increased H2AZK7me1 levels. In future experiments, we will explore the contribution of these RA-induced KMTs to H2AZ methylation.
Hints of Setd6 potential role in mESC self-renewal surfaced in a genetic screen of chromatin factors.Citation16 Our exhaustive investigations confirm, through AP staining (), immnunoblotting of Nanog (), competition () and clonogenic assays (), as well as cell death assays (Fig. S6) and cell cycle profiling (Figs. S7 and S8), that Setd6 is required for mESC self-renewal. Since Setd6-depleted cells appear to differentiate, it will be interesting to investigate how Setd6 contributes to cell lineages and identify the types of differentiated cells that emerge from decreased Setd6 expression. Together with the ChIP analyses of H2AZ and H2AZK7me1 at Fgf5, FoxA2 and Hand2 promoters, these results suggest that Setd6 participates in the maintenance of mESC self-renewal by regulating the expression of differentiation markers such as Fgf5. In addition, the loss of both H2AZK7me1 and H3K27me3 from Fgf5 upon cellular differentiation implies that H2AZK7me1 could be involved with H3K27me3 in an epigenetic program that regulates the expression of lineage commitment genes.
Quite interestingly, although the global level of H2AZK7me1 increased upon RA-induced mESC differentiation, the level of H2AZK7me1 assessed at the promoters of Fgf5, FoxA2, and Hand2 decreased along with the H3K27me3 mark. Whether H2AZK7me1 is demethylated by a demethylase or evicted by a histone exchange complex remains to be uncovered. However, it is clear that H2AZK7me1 levels decrease at some promoters upon RA-induced differentiation. Genome-wide studies, such as H2AZ and H2AZK7me1 ChIPseq, may reveal more precisely their biological functions. In addition, the eventual identification of H2AZ- or H2AZK7me1-specific readers will undoubtedly help concretize the molecular mechanism of chromatin signaling by H2AZK7me1.
To conclude, our results demonstrate that SETD6 monomethylates H2AZ on lysine 7 and that H2AZ is methylated in vivo at lysines 4 and 7. Notably, the H2AZK4me1K7me1 mark is induced upon mESC cellular differentiation. Finally, we show that Setd6 is required for the maintenance of mESC self-renewal. Although H2AZK7me1 and H2AZK4me1K7me1 increased in differentiated mESC, the self-renewal defects we observed upon silencing of Setd6 are likely due to a combination of lysine methylation signaling events, including H2AZK7me1 and H2AZK4me1K7me1 as well as other SETD6 substrates, such as the NF-κB subunit, RelA.Citation11 Future experiments should investigate the mechanism of H2AZK7me1 and H3K27me3 marks concomitant loss from promoters and how these histone marks may cooperate to regulate the expression of differentiation genes and maintain mESC self-renewal potential.
Materials and Methods
Plasmids, cDNA and peptides
H2AZ cDNA was cloned from HeLa cells by PCR on reverse transcribed total RNA using Pfu SuperMix and SuperScript II Reverse Transcriptase, respectively (Invitrogen; cat. no. 12532-016 and 18064-022, respectively). The indicated H2AZ cDNAs were inserted in either pGEX6-P-1 (GE Healthcare; cat. no. 12532-016) or pET28 (Novagen; cat. no. 69866-3) for bacterial expression. The H2AZ AGGKAGKme0/me1/me2/me3DSGKAKTKAVSRS-biotin and AGGKAGKme0/me1DSGKAKC peptides were synthesized at Yale University’s W.M. Keck Foundation Biotechnology Resource Laboratory.
Cell culture
U2OS and HEK293T cell lines were maintained in D-MEM (HyClone). The mouse ES NanogR rescue cell line was maintained as previously described.Citation12 E14 mESCs were maintained on 0.1% gelatin-coated tissue culture plates without feeder cells for all experiments. To induce differentiation with retinoic acid (RA), LIF (Chemicon; cat. no. ESG1107) was removed and RA (Sigma-Aldrich; cat. no. R2625) was added at a concentration of 5 μM. Alkaline phosphatase activity was measured using a commercial detection kit (Sigma-Aldrich).
Antibodies
The AGGKAGKme0/me1DSGKAKC peptides were conjugated to KLH (Sigma) according to the manufacturer’s suggested procedure, and used to immunize rabbits (Pocono Rabbit Farm and Laboratory, PA). An exhaustive list of the antibodies used can be found in the supplementary information. Full methods and associated references are available in the supplementary methods online.
| Abbreviations: | ||
| H2AZK7me1 | = | histone H2AZ monomethylated on lysine 7 |
| H3K27me3 | = | histone H3 tri-methylated on lysine 27 |
| KMT | = | lysine methyltransferase |
| SET | = | Su(var)3-9, enhancer-of-zeste, trithorax |
| SETD6 | = | SET domain-containing protein 6 |
| mESC | = | mouse embryonic stem cells |
| RA | = | retinoic acid |
| PRC2 | = | polycomb repressive complex 2 |
| SUZ12 | = | suppressor of zeste 12 |
| EED | = | embryonic ectoderm development |
| GST | = | glutathionine S-transferase |
| ChIP | = | chromatin immunoprecipitation |
Additional material
Download Zip (23.2 MB)Acknowledgments
The library screen was initiated in the laboratory of Or Gozani at Stanford University. This work was supported by Canadian Institute of Health Research (CIHR) grants (MOP-93811 and MOP-67070) and Weekend to End Women's Cancers grant from the Jewish General Hospital to SR, National Science Foundation (CBET-0941143) and a National Institutes of Health (NIH) grants (DP2OD007447) to BAG, and NIH (5R01GM078465) grant to IRL. O.B. held a postdoctoral fellowship from the CIHR and was supported by the McGill Integrated Cancer Research Training Program (FRN53888) while at McGill University. O.B. is supported by the Newcastle’s Biomedical Fellowship Programme, which is in part funded by the Wellcome Trust’s Institutional Strategic Support Fund.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/23416
References
- Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem Sci 2002; 27:396 - 402; http://dx.doi.org/10.1016/S0968-0004(02)02141-2; PMID: 12151224
- Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol 2010; 21:209 - 20; http://dx.doi.org/10.1016/j.semcdb.2009.10.007; PMID: 19892027
- Rahman N. Mechanisms predisposing to childhood overgrowth and cancer. Curr Opin Genet Dev 2005; 15:227 - 33; http://dx.doi.org/10.1016/j.gde.2005.04.007; PMID: 15917196
- Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009; 460:287 - 91; http://dx.doi.org/10.1038/nature08086; PMID: 19483677
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, et al. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc Natl Acad Sci U S A 2006; 103:19176 - 81; http://dx.doi.org/10.1073/pnas.0606373103; PMID: 17142323
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823 - 37; http://dx.doi.org/10.1016/j.cell.2007.05.009; PMID: 17512414
- Valdés-Mora F, Song JZ, Statham AL, Strbenac D, Robinson MD, Nair SS, et al. Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res 2012; 22:307 - 21; http://dx.doi.org/10.1101/gr.118919.110; PMID: 21788347
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, et al. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 2008; 135:649 - 61; http://dx.doi.org/10.1016/j.cell.2008.09.056; PMID: 18992931
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006; 441:349 - 53; http://dx.doi.org/10.1038/nature04733; PMID: 16625203
- Illingworth RS, Botting CH, Grimes GR, Bickmore WA, Eskeland R. PRC1 and PRC2 are not required for targeting of H2A.Z to developmental genes in embryonic stem cells. PLoS One 2012; 7:e34848; http://dx.doi.org/10.1371/journal.pone.0034848; PMID: 22496869
- Levy D, Kuo AJ, Chang Y, Schaefer U, Kitson C, Cheung P, et al. Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat Immunol 2011; 12:29 - 36; http://dx.doi.org/10.1038/ni.1968; PMID: 21131967
- Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, et al. Dissecting self-renewal in stem cells with RNA interference. Nature 2006; 442:533 - 8; http://dx.doi.org/10.1038/nature04915; PMID: 16767105
- Bruce K, Myers FA, Mantouvalou E, Lefevre P, Greaves I, Bonifer C, et al. The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res 2005; 33:5633 - 9; http://dx.doi.org/10.1093/nar/gki874; PMID: 16204459
- Lee D-F, Su J, Sevilla A, Gingold J, Schaniel C, Lemischka IR. Combining competition assays with genetic complementation strategies to dissect mouse embryonic stem cell self-renewal and pluripotency. Nat Protoc 2012; 7:729 - 48; http://dx.doi.org/10.1038/nprot.2012.018; PMID: 22441292
- Lee D-F, Su J, Ang Y-S, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF, et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell 2012; 11:179 - 94; http://dx.doi.org/10.1016/j.stem.2012.05.020; PMID: 22862944
- Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev 2009; 23:2484 - 9; http://dx.doi.org/10.1101/gad.1837309; PMID: 19884255
- Draker R, Sarcinella E, Cheung P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res 2011; 39:3529 - 42; http://dx.doi.org/10.1093/nar/gkq1352; PMID: 21245042
- Sarcinella E, Zuzarte PC, Lau PNI, Draker R, Cheung P. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol Cell Biol 2007; 27:6457 - 68; http://dx.doi.org/10.1128/MCB.00241-07; PMID: 17636032