Abstract
DNA methylation, an important type of epigenetic modification in humans, participates in crucial cellular processes, such as embryonic development, X-inactivation, genomic imprinting and chromosome stability. Several platforms have been developed to study genome-wide DNA methylation. Many investigators in the field have chosen the Illumina Infinium HumanMethylation microarray for its ability to reliably assess DNA methylation following sodium bisulfite conversion. Here, we analyzed methylation profiles of 489 adult males and 357 adult females generated by the Infinium HumanMethylation450 microarray. Among the autosomal CpG sites that displayed significant methylation differences between the two sexes, we observed a significant enrichment of cross-reactive probes co-hybridizing to the sex chromosomes with more than 94% sequence identity. This could lead investigators to mistakenly infer the existence of significant autosomal sex-associated methylation. Using sequence identity cutoffs derived from the sex methylation analysis, we concluded that 6% of the array probes can potentially generate spurious signals because of co-hybridization to alternate genomic sequences highly homologous to the intended targets. Additionally, we discovered probes targeting polymorphic CpGs that overlapped SNPs. The methylation levels detected by these probes are simply the reflection of underlying genetic polymorphisms but could be misinterpreted as true signals. The existence of probes that are cross-reactive or of target polymorphic CpGs in the Illumina HumanMethylation microarrays can confound data obtained from such microarrays. Therefore, investigators should exercise caution when significant biological associations are found using these array platforms. A list of all cross-reactive probes and polymorphic CpGs identified by us are annotated in this paper.
Introduction
There has been a recent surge of epigenetic studies targeting a large group of biological phenomena, ranging from the fundamental subject of cell cycle regulation to the interdisciplinary topic of socio-economic position.Citation1,Citation2 Epigenetic mechanisms include, but are not limited to, DNA methylation, histone modification and microRNAs. Of these, DNA methylation is the most readily accessible for scientific research, because DNA can be easily extracted with the methyl group firmly anchored to the 5′ site of the cytosine ring in CG dinucleotides (CpGs). Gene expression is known to be directly regulated by DNA methylation and DNA methylation alterations can lead to disease phenotypes.Citation3 Furthermore, DNA methylation has been demonstrated to be an important surrogate for other epigenetic changes relevant to normal biological processes, as well as genetic defects, or environmental influence.Citation4 Thus, DNA methylation represents a valuable and indispensible tool for understanding human biology.
The Illumina Infinium HumanMethylation450k microarray is one of the most comprehensive microarray platforms available for the study of genome-wide DNA methylation in humans. It assesses the methylation levels of 485,577 CpG sites, covering 99% of RefSeq genes and all the different epigenetically important genomic regions such as CpG island, island shore and shelf, 5′ and 3′ UTRs, and promoter and gene body. This microarray platform works similarly to the Illumina SNP microarrays.Citation5 It generates quantitative genotypes of bisulfite-treated CpG sites instead of SNPs. About one third of the CpGs are interrogated by the Infinium I probes and the rest by the Infinium II probes. The Infinium I and II probes are both 50 bases long but detect methylation levels by slightly different mechanisms. The paired probe approach of the Infinium I technology uses two probes designed for each CpG, one for the methylated and the other for the unmethylated sequence. Essentially, the two probes in each pair differ at the end-nucleotide that matches to the cytosine position of a CpG. The end-nucleotide of the probe can be either a guanine complementing the cytosine of the methylated CpG, or an adenine complementing the thymine resulting from the bisulfite transformation of the unmethylated cytosine. The two probes can differ at sites other than the end-nucleotide if there are additional CpGs within the 50-base hybridization sequence. The fluorescent-labeled single base extension, which occurs only upon correct hybridization of the probe’s end-nucleotide to the CpG site, provides the array signals. The methylation level is determined from the differential signal intensities detected by the two probes. On the other hand, the Infinium II technology utilizes the single probe two-color approach that relies on red and green fluorescent-labeled single base extension occurring differentially at thymine and cytosine of bisulfite converted DNA. The methylation level is determined based on the differential signal intensities detected by the two-color channels.
We report our findings of cross-reactive probes in the Illumina 450k microarray based on array methylation profiles of 489 males and 357 females. These cross-reactive probes target repetitive sequences or co-hybridize to alternate sequences that are highly homologous to the intended targets and thus could generate spurious signals, potentially resulting in invalid conclusions and lack of validation in downstream analyses. Also of importance is our finding of probes targeting CpG sites that overlap known SNPs or what we referred to as polymorphic CpGs. The methylation levels detected for such CpGs can be greatly influenced by the underlying genetic polymorphisms; thus they should be interpreted with caution.
Results
Probe cross-reactivity can lead to the identification of spurious autosomal sex-associated methylation differences
We analyzed data from the Illumina 450k methylation profiles of 489 males and 357 females from the control cohort of the Assessment of Risk for Colorectal Cancer Tumours in Canada (ARCTIC) project. We identified 16,532 autosomal CpGs with significant sex-associated methylation differences (Bonferroni-corrected p value < 0.05). This set is fairly robust to the choice of data normalization method: using SWAN,Citation6 the overlap is over 96%. In order to assess the effect of cross-reactivity on the observed sex methylation differences, we first searched for potential cross-reactive targets by mapping all the 473,864 autosomal probes to the sex chromosomes of in silico bisulfite-converted reference genomes (build 37; hg19). By plotting the frequency of mapped matches representing potential cross-reactive targets, we found significant enrichments of high identity matches among the autosomal probes displaying significant sex-associated methylation differences compared with those detecting no significant differences (). Specifically, enrichments were observed for the Infinium I and II autosomal probes with sex-chromosomal matches that had at least 47 bases sequence identity (enrichment p value: 2.36E-06 for Infinium I and 1.67E-03 for Infinium II). We further analyzed the same data using additional criteria such as the number of cross-reactive targets. We found that even with a single cross-reactive target of 47 bases, the significance of the enrichment remained (p-value: 2.30E-04 for Infinium I and 5.43E-04 for Infinium II). The two Infinium type probes were analyzed separately because of the different chemistries involved in measuring methylation levels. The paired probes approach of Infinium I should theoretically be more sensitive to the differential methylation effect of cross-reactivity, because probes detecting the methylated and the unmethylated targets could be quite different due to within-probe CpG sites hybridized by CG in one probe and CA in the other probe. Such a difference does not exist in Infinium II probes, since one probe is designed to target both methylated and unmethylated targets with degenerate probe sequences (R nucleotide for G/A). We showed several sex-specific distributions of methylation levels detected by autosomal probes co-hybridizing to sex chromosomes, exemplifying the effect of cross-reactivity (; Figs. S1 and S2A). We suggest that significant findings based on data generated by these cross-reactive probes are likely to be technical artifacts rather than true biological phenomena. We further proposed the minimum number of bases matched to unintended targets of 47 bases for Infinium I and II probes to be used as criteria to identify cross-reactivity. By mapping all array probes against the in silico bisulfite-converted reference genomes, we found 8.4% of the Infinium I probes and 5.1% of the Infinium II probes (or 6.0% of total probes) to be cross-reactive ().
Figure 1. Enrichment of high identity matches on sex chromosomes for autosomal-targeting probes with significant sex methylation differences. (A) Distribution of Infinium I and (B) Infinium II probes mapped to the sex chromosomes. BLAT was performed on autosomal probes against the in silico bisulfite-converted sex chromosomes. Only the best BLAT match for each probe was used to determine the number of bases that matched (x-axis). The significance of sex-associated methylation difference is based on methylation profiles derived from ARCTIC control cohorts.
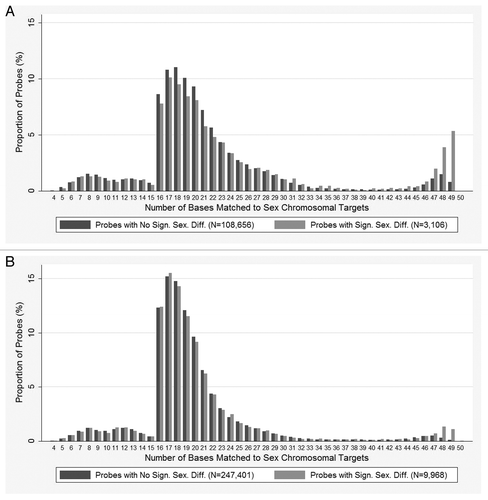
Figure 2. Methylation profile graph of SEPHS1P locus where several probes map onto chromosome Y and one probe targets a polymorphic CpG. The colored bars represent the methylation profile across all controls, females on the left of the dashed line, male on the right. The bottom dot plot shows the significance of the male/female methylation differences (open circles are p-values < 10^-12). All probes with p-values < 10^-12 map onto chromosome Y. cg05455372 targets a polymorphic CpG cytosine (rs2863984), which has an allele frequency of 0.42 according to 1000 Genomes data. The schematic representations of probes targeting cg04462931 and cg05455372 are shown in Figure S2.
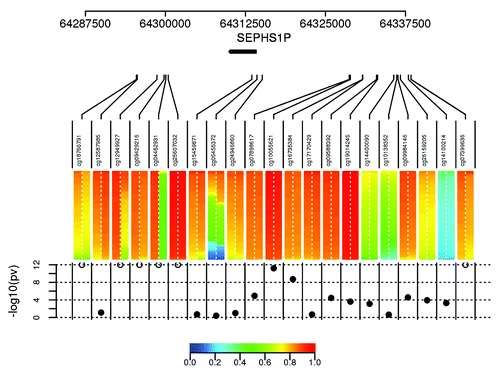
All probes were mapped to intended targets with perfect matches at the correct genomic strand and location coordinate, suggesting successful sequence mapping except for probes targeting non-CpG cytosine. Of the 3,091 non-CpG targeting probes (Probe ID: ch.[..].[.....]), which we excluded from the overall calculation of cross-reactive probes in , only about one third can be mapped with a perfect match to the correct genomic location annotated by the Illumina ().
Methylation data of polymorphic CpGs reflect underlying genetic polymorphism
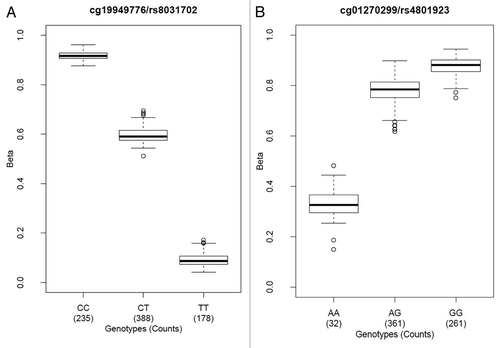
In addition to cross-reactive probes, we describe a subset of CpGs that overlap known SNPs (i.e., CpGs that are polymorphic at cytosine or guanine positions). Of these polymorphic CpGs, appropriate interpretation of methylation data requires a priori knowledge of each individual’s genotype. Also of importance is the “base before CpGs” for the Illumina I probes, because that is where the signal-generating single base extension occurs. By cross-matching the genomic positions of both C and G of all array-targeted CpGs and the position of single base extension (Infinium I) to that of known SNPs in the 1000 Genome database, we found 9.4% of the Infinium I probes and 15.5% of the Infinium II probes (13.8% of total probes) to have methylation levels that could potentially be affected by genetic polymorphisms (). By utilizing the genotyping data of the same individuals, we observed methylation profiles that could be explained by patterns of SNP genotypes ( and ; Figs. S2B and S3). Although we showed that methylation data from this microarray could be greatly affected by genetic polymorphism, the majority of these SNPs are rare with very low alternative allele frequencies (Fig. S4). Thus, they would not be expected to have a major effect on the methylation data when the population under study does not demonstrate a significant frequency of the rare allele.
Figure 3. Polymorphic CpG methylation can reflect underlying SNP genotypes. (A) C/T SNP located at the cytosine position of a polymorphic CpG targeted by an Infinium II probe. The C allele was detected as a methylated allele, while the T allele was detected as an unmethylated allele. (B) A/G SNP located at the guanine position of a polymorphic CpG targeted by an Infinium I probe. The A allele transforms the locus into a non-CpG, which on one hand can lead to true loss of methylation and on the other hand can prohibit single base extension from occurring due to loss of hybridization at the SNP location. The low detection count of methylation calls for the AA genotype was the result of loss of hybridization leading to low signal intensity.
Moreover, we found that 239,238 sites (49.3% of all sites) have a probe that overlaps at least one SNP, including 85,771 sites (17.7%) for which the SNP's non-reference allele has a frequency of at least 1% and 55,666 sites (11.5%) where the allele frequency is at least 5% (allele frequency estimated from all 1000 Genome project samples). For 80,717 CpG sites (16.6%), the SNP is located within 10 bases of the query site where single-base extension occurs, with an allele frequency of at least 1% for 19,418 sites (4.0%) and an allele frequency of at least 5% for 10,825 sites (2.2%).
The list of polymorphic CpGs targeted by the array and the list of SNPs underlying probe hybridization sequences are available as Supplemental Data.
Discussion
In this paper, we report that 6% of the Illumina 450k microarray probes are cross-reactive, co-hybridizing to alternate sequences highly homologous to the intended targets. This is comparable to the 6–10% of cross-reactive probes we previously reported for the Illumina 27k microarray.Citation7 We have previously shown that the cross-reactivity is primarily the result of probes targeting repetitive genomic sequences or genes that have pseudogenes or homologous genes. The cross-reactive sites could reflect CpGs of different methylation status or non-CpGs that are detected as fully methylated or unmethylated loci. The cross-reactive probes were originally discovered when investigating sex-associated DNA methylation on autosomes. We found that the top candidate CpGs were targeted by probes with sequences that also mapped to the sex chromosomes with high identity matches. The observed methylation differences were thus attributable to the methylation and copy number differences of the sex chromosomes in normal males vs. females. One copy of the female X chromosome is heavily methylated due to X-inactivation while only males have a Y chromosome. For female X chromosome, the methylation can appear skewed to either sex depending on whether the cross-reactive target is on the methylated inactive X chromosome or on the unmethylated active X chromosome. Since only females have the heavily methylated X chromosome, we observed the expected, that there were more cross-reactive probes detecting higher female methylation than those detecting higher male methylation (830 sites vs. 258 sites). To our knowledge, four previous peer-reviewed papersCitation8-Citation11 used data from the Illumina 27k microarray to report the same overlapping set of autosomal sex-associated methylation differences, which we found to be technical artifacts created by autosomal probes cross-reacting with genomic regions on the sex chromosomes.Citation7 Such false discovery could have been avoided if an independent validation of the microarray findings by a second method, such as bisulfite-pyrosequencing, had been undertaken.
Also of concern are the polymorphic CpGs targeted by the Illumina 450k microarray. We demonstrated that the methylation data at these polymorphic CpGs reflect the underlying genetic polymorphism. We suspect that these polymorphic CpGs will have the greatest impact on the interpretation of findings of methylation quantitative trait loci (mQTLs), especially in the case in which cis-associated SNPs occur at the same haplotype as the polymorphic CpGs. That is, the methylation variation observed in mQTLs is representative not of the association between methylation levels and SNPs but of the linkage disequilibrium that exists between polymorphic CpGs and associated SNPs. Alternatively, cross-reactive probes that unintentionally target in trans-associated SNPs or polymorphic CpGs in close linkage of trans-associated SNPs can lead to findings of trans-mQTLs. Besides mQTLs, any quantitative association between a variable of interest and methylation at these polymorphic CpGs cannot be ascertained unless the confounding effect of the genetic polymorphism can be addressed by independent methods such as SNP genotyping microarray. Note that studies that are focused on intraindividual differences rather than interindividual differences (for example, tumor/normal tissue differences; longitudinal evaluation of methylation profiles; monozygotic twin studies) are not expected to be confounded by such underlying SNPs.
In summary, we recommend cautious interpretation of microarray data with special emphasis on potential signals generated by cross-reactive probes and polymorphic CpGs. Users of the Illumina 450k microarray should cross-check their candidate CpGs using the list of cross-reactive probes and polymorphic CpGs that we have made publicly available to the scientific community (Supplemental Data). In addition, we also made available the list of SNPs underlying the probe hybridization sequences as an update to the Illumina annotation. Candidate CpGs targeted by cross-reactive probes should be validated by a second independent approach, whereas candidates involving polymorphic CpGs should have potential underlying SNPs genotyped. In this way, false interpretation of technical artifacts generated by cross-reactivity and the biological artifacts secondary to genotypic polymorphisms can be avoided. Other quality control procedures, such as peak correctionCitation12 and array normalization,Citation13 should also be cautiously considered with a view to generating high quality genome-wide DNA methylation data and meaningful biological conclusions.
Methods
Methylation profiling
We profiled the epigenetic landscape of 990 unique donors forming the control cohort of the Assessment of Risk for Colorectal Cancer Tumours in Canada (ARCTIC) project.Citation14 Fifteen μl of lymphocyte-derived DNA extracted (at an average concentration 90 ng/μl) was bisulfite-converted using the EZ-96 DNA Methylation-Gold Kit (Zymo Research, Orange, CA); 4μl of bisulfite-treated DNA was then analyzed on the HumanMethylation450 BeadChip from Illumina according to the manufacturer’s protocol. Intensities were normalized using Illumina’s internal normalization probes and algorithms, without background subtraction. Beta values with assigned detection p-values > 0.01 were treated as missing data. CpG sites with more than 1% missing data across all samples were discarded.
We removed from analysis samples that were outliers with respect to any one of the internal control probes (excluding probes designed to evaluate the background noise and probes designed to normalize the data) and samples that were not of non-Hispanic white ancestry, either self-declared or by investigation of genetic ancestry using genome-wide SNP data. After sample exclusion, we were left with 489 adult males and 357 adult females.
Autosomal sex methylation analysis
For each site, we evaluated the significance of the differences in methylation levels between males and females using a linear model, where the dependent variable is the β value after logit transformation and sex is the independent variable. The model was adjusted for the following covariates: the age of the individual, an indicator for the array on which the DNA sample was processed and an indicator of the position (row and column) on the array where the sample was found. In addition, we used a 2-sample test for equality of proportions with continuity correction to determine the significance of enrichment in sex-associated methylation differences for probes with high sequence identity to sex chromosomes.
Mapping cross-reactive probes
The human reference genome build 37 (hg19) was downloaded from UCSC genome browser. Both strands of the reference genome were bisulfite-converted separately in silico to represent the unmethylated and methylated genomes post-bisulfite conversion. In both genomes, all Cs of non-CpG sites are converted to Ts; in the unmethylated genome, all Cs of CpG sites are also converted to Ts, whereas in the methylated genome, all Cs of CpG sites remain as Cs. A total of 4 non-complementary single-stranded genomes (forward methylated, forward unmethylated, reverse methylated, reverse unmethylated) were generated to represent all possibilities post-bisulfite conversion, and subsequently the sequence-mapping program, BLAT, internally generated the other 4 single-stranded genomes that are complement to the 4 in silico bisulfite-converted single-stranded genomes.
The probe sequence of the Infinium I probes was easily extracted from the annotated file provided by the Illumina. There are two probe sequences for each Infinium I targeted CpG sites, whereas there is only one probe sequence for each Infinium II targeted CpG sites. For Infinium II, some probe sequences in the annotated file contain R nucleotide, representing either A or G due to the presence of CpG sites within the probe sequence. Here, the A would match to T of unmethylated CpG, and the G would match to C of methylated CpG. For these probes, we generated all possible probe sequences by replacing all R nucleotides with all possible combinations of A and G nucleotides. In the end, 1,119,246 probe sequences were obtained for all array probes.
All probe sequences were mapped against the 8 single-stranded bisulfite-converted reference genomes using BLAT.Citation15 The BLAT parameter used was -stepSize = 5 -repMatch = 10000000000 -minScore = 0 -minIdentity = 0 -maxIntron = 0. Only matches with end-nucleotide match to the probe sequences were retained, because end-nucleotide match is necessary to generate array signals and thus to have any cross-reactive effect. Duplicate matches of the same probe that map to the same chromosomal location were removed, and only the one with the highest sequence identity was retained. Matches with gaps were also removed, since gaps could significantly reduce the degree of cross-reactivity. For probes targeting CpG sites in regions for which alternative assemblies exist (e.g., chr4_ctg9_hap1), matches to the corresponding alternative assemblies were removed to avoid double-counting the same match on the primary (e.g., chromosome 4) and alternative loci assemblies. For autosomal sex methylation analysis, only matches of the autosomal-targeting probes that mapped to the sex chromosomes were retained. To generate a list of cross-reactive probes, all matches are filtered based on one additional criterion, the total number of bases matched (47 bases for both Infinium I and II), derived from the sex methylation analysis.
Identification of polymorphic CpGs
We interrogated the 20110521 release of the 1000 Genomes projectCitation16 to generate a list of CpG sites that are potentially polymorphic. A CpG site was deemed to be polymorphic if a SNP resided at the position of the cytosine or guanine on either strand, and, in the case of Infinium I assays, if a SNP resided at the position where single base extension occurs (base before C). Allele frequencies were extracted. We also looked for polymorphic sites within each probe target sequence. To illustrate the methylation profiles of polymorphic CpGs, we used SNP data described elsewhere (a 1536 GoldenGate panel from Illumina; the 10k coding-SNP array from Affymetrix/ParAllele; the Human Mapping 100k set from Affymetrix;Citation14) in addition to SNPs genotyped with the Human Mapping 500k set from Affymetrix.
| Abbreviations: | ||
| CpGs | = | CG dinucleotides |
| SNPs | = | single nucleotide polymorphisms |
Additional material
Download Zip (630 KB)Acknowledgments
The methylation profiling of the OFCCR samples was funded by a GL2 grant from the Ontario Research Fund to B.W.Z. and T.J.H. B.W.Z. and T.J.H. are recipients of Senior Investigator Awards from the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Economic Development and Innovation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Borghol N, Suderman M, McArdle W, Racine A, Hallett M, Pembrey M, et al. Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol 2012; 41:62 - 74; http://dx.doi.org/10.1093/ije/dyr147; PMID: 22422449
- Shen L, Toyota M, Kondo Y, Obata T, Daniel S, Pierce S, et al. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood 2003; 101:4131 - 6; http://dx.doi.org/10.1182/blood-2002-08-2466; PMID: 12586619
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999; 21:163 - 7; http://dx.doi.org/10.1038/5947; PMID: 9988266
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33:Suppl 245 - 54; http://dx.doi.org/10.1038/ng1089; PMID: 12610534
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics 2009; 1:177 - 200; http://dx.doi.org/10.2217/epi.09.14; PMID: 22122642
- Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 2012; 13:R44; http://dx.doi.org/10.1186/gb-2012-13-6-r44; PMID: 22703947
- Chen YA, Choufani S, Ferreira JC, Grafodatskaya D, Butcher DT, Weksberg R. Sequence overlap between autosomal and sex-linked probes on the Illumina HumanMethylation27 microarray. Genomics 2011; 97:214 - 22; http://dx.doi.org/10.1016/j.ygeno.2010.12.004; PMID: 21211562
- Liu J, Morgan M, Hutchison K, Calhoun VD. A study of the influence of sex on genome wide methylation. PLoS One 2010; 5:e10028; http://dx.doi.org/10.1371/journal.pone.0010028; PMID: 20386599
- Adkins RM, Thomas F, Tylavsky FA, Krushkal J. Parental ages and levels of DNA methylation in the newborn are correlated. BMC Med Genet 2011; 12:47; http://dx.doi.org/10.1186/1471-2350-12-47; PMID: 21453505
- Adkins RM, Krushkal J, Tylavsky FA, Thomas F. Racial differences in gene-specific DNA methylation levels are present at birth. Birth Defects Res A Clin Mol Teratol 2011; 91:728 - 36; http://dx.doi.org/10.1002/bdra.20770; PMID: 21308978
- Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet 2012; 90:260 - 72; http://dx.doi.org/10.1016/j.ajhg.2011.12.020; PMID: 22305529
- Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011; 3:771 - 84; http://dx.doi.org/10.2217/epi.11.105; PMID: 22126295
- Touleimat N, Tost J. Complete pipeline for Infinium(®) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012; 4:325 - 41; http://dx.doi.org/10.2217/epi.12.21; PMID: 22690668
- Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 2007; 39:989 - 94; http://dx.doi.org/10.1038/ng2089; PMID: 17618283
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res 2002; 12:656 - 64; PMID: 11932250
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al, 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061 - 73; http://dx.doi.org/10.1038/nature09534; PMID: 20981092