Abstract
Myogenic cell cultures derived from muscle biopsies are excellent models for human cell differentiation. We report the first comprehensive analysis of myogenesis-specific DNA hyper- and hypo-methylation throughout the genome for human muscle progenitor cells (both myoblasts and myotubes) and skeletal muscle tissue vs. 30 non-muscle samples using reduced representation bisulfite sequencing. We also focused on four genes with extensive hyper- or hypo-methylation in the muscle lineage (PAX3, TBX1, MYH7B/MIR499 and OBSCN) to compare DNA methylation, DNaseI hypersensitivity, histone modification, and CTCF binding profiles. We found that myogenic hypermethylation was strongly associated with homeobox or T-box genes and muscle hypomethylation with contractile fiber genes. Nonetheless, there was no simple relationship between differential gene expression and myogenic differential methylation, rather only for subsets of these genes, such as contractile fiber genes. Skeletal muscle retained ~30% of the hypomethylated sites but only ~3% of hypermethylated sites seen in myogenic progenitor cells. By enzymatic assays, skeletal muscle was 2-fold enriched globally in genomic 5-hydroxymethylcytosine (5-hmC) vs. myoblasts or myotubes and was the only sample type enriched in 5-hmC at tested myogenic hypermethylated sites in PAX3/CCDC140 andTBX1. TET1 and TET2 RNAs, which are involved in generation of 5-hmC and DNA demethylation, were strongly upregulated in myoblasts and myotubes. Our findings implicate de novo methylation predominantly before the myoblast stage and demethylation before and after the myotube stage in control of transcription and co-transcriptional RNA processing. They also suggest that, in muscle, TET1 or TET2 are involved in active demethylation and in formation of stable 5-hmC residues.
Introduction
The importance of tissue-specific differences in human DNA methylationCitation1 is emerging with much greater clarity because of the recent availability of human methylome profilesCitation2 and the recognition of 5-hydroxymethylcytosine (5-hmC) as the sixth genetically programmed base in mammalian DNA.Citation3 This modified base appears to be a functional component of the epigenome as well as an intermediate in the conversion of certain genomic 5-methylcytosine (5-mC) residues to C residues.Citation4-Citation6 DNA methylome analysis can be used to study differentiation- or disease-linked differences in human epigenetics from a wide range of tissues and cell types more readily than can chromatin epigenetic profiling. Moreover, it gives insights into chromatin epigenetics, with which it is often coupled, albeit sometimes in complex relationships.Citation7
The differentiation of mononuclear myoblasts (Mb) to multinuclear myotubes (Mt) provides an in vitro model for complex differentiation-linked changes in cellular physiology that are critical during embryogenesis and postnatal, regenerative muscle repair. The muscle lineage-specific hyper- and hypo-methylation throughout the genome has not been examined, although there have been studies of promoter methylation in skeletal muscleCitation8-Citation10 or myoblasts vs. adipose cell-derived myocytes.Citation11 Therefore, by reduced representation bisulfite sequencing (RRBS),Citation12 we profiled DNA methylation in untransformed Mb cultures and Mt samples derived from them as well as in 16 types of cell cultures established from normal human tissues in order to identify myogenesis-associated changes in DNA methylation. In addition, we generated analogous profiles from skeletal muscle and diverse non-muscle tissues. The Mb preparations used for this study retain the ability to efficiently differentiate into Mt, and the Mb had the expected specificity of their expression profiles,Citation13 indicating that they largely maintain their in vivo characteristics.
RRBS has single-base resolution and detects CpGs in gene bodies as well as intergenic regions, CpG island (CGI) and non-island sequences, and both single-copy and repeated sequences, even though only ~5% of genomic CpGs are detected by this procedure. By sampling only a fraction of CpG sites, but including all the major classes of DNA sequences, RRBS enables comparison of methylomes from many different samples. Because standard bisulfite-based techniques to study DNA methylation, like RRBS, are generally unable to distinguish genomic 5-mC that is 5-hmCCitation14 from that which is just methylated, we also determined the total genomic 5-hmC content of myogenic progenitor cells, skeletal muscle, and several types of non-muscle cell cultures or tissues and differentiated 5-hmC from 5-mC in several key gene regions. With the major exception of brain, there is usually less than 5% as much 5-hmC as 5-mC in human tissues.Citation14-Citation16
We report many novel findings about myogenesis- and muscle-associated epigenetics from our comparison of the methylomes of an unusually large number of various human samples (32 cell cultures and 16 tissue samples) and from enzymatic assays for genomic 5-hmC. Hypermethylation in Mb and Mt showed a very strong association with genes encoding sequence-specific transcription factors, including homeodomain and T-box proteins. The vast majority of this hypermethylation was lost in skeletal muscle. Hypomethylation in skeletal muscle had very strong associations with genes encoding muscle-specific proteins, and these were much stronger than those for hypomethylation in Mb and Mt. To better understand the biological context of myogenic hyper- and hypo-methylation, we examined in detail the locations of differentially methylated (DM) sites and myogenesis-associated chromatin epigenetic marks (DNaseI hypersensitivity, histone modification and CTCF binding) for four genes that were associated with 20 to 131 DM sites. We also looked for correlations with expression of these genes that previously had no description of their DNA methylation status or none of their methylation outside the promoter region. Two of these genes displaying myogenic hypermethylation, PAX3 (a homeobox gene) and TBX1 (a T-box gene), encode transcription factors involved in various developmental pathways, including myogenesis.Citation17-Citation19 PAX3 is associated with the craniofacial-deafness-hand syndrome, the Waardenburg syndrome, and rhabdomyosarcoma andTBX1 with the DiGeorge 22q11.2 deletion syndrome and conotruncal heart defects.Citation17,Citation19,Citation20 In tested myogenic hypermethylated sites associated with these genes, we found that skeletal muscle was the only sample enriched in 5-hmC. The other two highlighted genes, MYH7B/MIR499 and OBSCN, which exhibited mostly muscle-lineage hypomethylation, are important in the formation of skeletal muscle as well as in certain other types of organogenesis and are implicated in cardiomyopathy.Citation21-Citation23 Our study reveals the different timing of de novo methylation and demethylation during muscle formation, indicates the importance of differentially methylated sites in centrally located exons and introns in the muscle lineage, and provides new insights into epigenetic changes in genes that are clinically or developmentally important.
Results
Hyper- and hypo-methylation in myogenic progenitors and muscle vs. non-muscle samples
We compared RRBS DNA methylation profiles from immunocytochemically characterized skeletal muscle progenitor cell cultures (nine Mb and Mt preparations) with those of 15 different types of non-transformed human cell strains plus Epstein-Barr virus-transformed lymphoblastoid cell lines (LCLs; Table S1). For each sample type, 2 to 8 biological or technical replicates were included. The Mb cultures (~70% confluent) were derived from quadriceps biopsies of two control individuals, 27 y, F, and 46 y, M), one patient with inclusion body myositis (IBM; 74 y, F), and gastrocnemius or quadriceps from two genetically confirmed FSHD (an autosomal dominant disease) patients (29 y, M, and 14 y, F). Mb cultures were differentiated to Mt samples (> 70% of nuclei in multinucleated, desmin-positive cells) by serum limitation. The inclusion of the IBM Mb sample provided a disease control for FSHD myogenic progenitor cells and a relatively rare myoblast sample from an older individual for comparison to the skeletal muscle samples from individuals of similarly advanced ages. Our previous expression profiling on exon-based microarrays of normal-control, IBM, and FSHD Mb and Mt samplesCitation13 indicated that these samples had mostly similar, myogenesis-specific expression profiles, although there were significant differences between the FSHD and control myogenic samples. However, these were usually only in the extent of myogenesis-associated changes in gene expression.
The number of RRBS sites per sample ranged from about 1 to 2 million. Approximately 50% of these sites should overlap CGIs.Citation12 Some of the sites appearing to be 5-mC in RRBS data sets might be 5-hmC; however, in most cell types other than brain, the amount of C methylation vastly outnumbers C hydroxymethylation.Citation14 Therefore, we use the term “DNA methylation” to include both 5-mC and 5-hmC. We compared the Mb and Mt samples as a group (MbMt) to the non-muscle cell types because the differences between Mb and Mt were very much less than between these samples and non-muscle cell cultures. Combining them greatly increased our statistical power for detecting myogenic DM sites. Stringent criteria were set for determining DM sites in the MbMt data set vs. the other cell cultures, namely, at least a 50% difference in methylation in the myogenic cells vs. the other cell cultures at a significance level of p < 0.01, as determined by fitted binomial regression models at each monitored CpG site. With these criteria, 19640 sites (1.7% of those assayed that had sufficient coverage) exhibited significant myogenesis-associated methylation. There was strong overrepresentation of myogenesis-relevant functional group terms for nearby genes, as described below. In addition, we examined RRBS profiles of two skeletal muscle samples (referred to as “muscle”; 83 y, F, and 71 y, M, with technical duplicates) vs. those from 12 different types of normal, non-muscle tissues plus two types of short-term, primary cell cultures (pancreatic islets and hepatocytes).
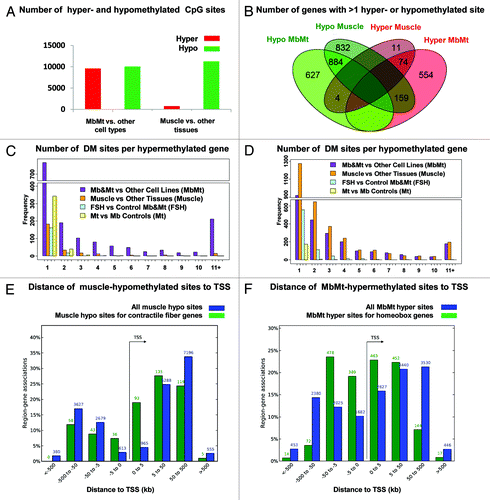
Of the total MbMt-associated DM sites, 9592 (49%) were hypermethylated vs. non-muscle cell cultures and 10048 (51%) were hypomethylated (). In contrast, the distribution of the 12,016 DM sites in skeletal muscle vs. other tissues was highly skewed, with 761 sites (6%) hypermethylated and 11,255 sites (94%) hypomethylated. We mapped DM sites to the closest gene and then determined the overlap of genes that exhibited muscle and MbMt differential methylation. For this analysis we used genes containing at least two DM sites that were hypomethylated or two that were hypermethylated; genes with both hypo- and hyper-methylated DM sites were designated according to which type of DM site was in the majority. Although 91% of the genes with hypermethylation at the Mb and Mt stages did not display hypermethylation in muscle, many of the genes that were hypermethylated in muscle were also hypermethylated in Mb and Mt samples (74 of the 89 genes, 83%; ). This indicates that muscle hypermethylation consisted mostly of the small percentage of retained MbMt hypermethylation. In contrast, only 47% of the genes that were hypomethylated in muscle were also hypomethylated in the MbMt group (885 of 1,876 genes), indicating that much of the muscle hypomethylation was established after the Mt stage. Another difference between myogenic hyper- and hypo-methylation is that a high density of MbMt DM sites was more prevalent among the hypermethylated genes ( vs. D) and hypermethylated sites were much more frequently arrayed within 5 kb of the transcription start site (TSS) than were hypomethylated sites (, blue bars). That muscle tissue retained an even tighter distribution of hypermethylated sites around the TSS than did Mb and Mt (Fig. S1) suggests the importance of this hypermethylation in repressing or reinforcing repression of many homeobox genes selectively in the muscle lineage.
Figure 1. Distribution of myogenic hyper- and hypo-methylated sites. Genome-wide distribution of sites that were differentially methylated (DM) in myoblasts and myotubes vs. non-muscle cell cultures or in skeletal muscle vs. non-muscle tissues are shown. (A) Numbers of DM sites. (B) Venn diagram with overlap of genes associates with > 1 DM site using the majority type of DM site associated with a gene for its hyper- or hypomethylated classification. (C and D) number of hyper- and hypo-methylated sites per associated gene as determined by closestBed analysis. (E and F) distance of from the associated gene’s transcription start site (TSS) of all muscle-hypomethylated or MbMt-hypermethylated CpG sites (blue bars) or just DM sites from genes encoding contractile fiber proteins or genes with a conserved homeobox site as determined by GREAT analysis.
Functional and regional correlates of myogenic hypo- and hyper-methylation
To test for preferential distribution of DM sites in different subgenic regions, we mapped all the DM sites to the following regions with respect to a single RefSeq isoform for each gene: 2 kb upstream of the TSS (-2 kb) through the first intron, the last exon plus 2 kb downstream, the internal exons, the internal introns, and intergenic regions > 2 kb from the gene ends. About 34.5 and 45.5% of the MbMt and muscle DM sites were in internal exons or introns, respectively (Table S2B). When the ratio of the number of internal-exon to internal-intron DM sites was normalized for the average size of the exons and introns in these genes, there appeared to be a strong bias for myogenic differential methylation in exons (Table S2C). However, when the number of RRBS-detected CpG sites was used for the normalization, no genome-wide bias toward exons vs. introns was seen because RRBS had a strong preference for detecting CpGs in exons, which is likely to be of biological significance.Citation24
Genes were then divided into those with mostly myogenic hyper- or hypo-methylation and examined with the Genomic Regions Enrichment of Annotations Tool (GREATCitation25). This program uses proximity and functional data to map small epigenetically defined subregions or sites to the TSS of one or more nearby genes. The strongest associations between myogenic DM sites and functional terms for nearby genes were for MbMt-hypermethylated sites and genes encoding sequence-specific DNA binding proteins, in general, or homeobox and T-box transcription factors, in particular (Table S2A). There were 1,671 and 351 sites, respectively, in these functional groupings (FDR Q-values < 10−200). Because RRBS favors CGIs, we checked whether these associations were due to such a bias. This was not the case because upon analysis of only the myogenic DM sites that did not overlap a CGI, the above functional terms were still highly significantly overrepresented among MbMt-hypermethylated sites (data not shown). Moreover, there was a strong association of histone genes with CGIs but no association of myogenic hypermethylation with histone genes. In addition, T-box genes were not associated with CGIs despite their strong association with MbMt-hypermethylated DM sites. Furthermore, the association of myogenic hypermethylation with homeobox genes was much stronger than that for CGIs. The highly significant associations of the promoters of muscle hypomethylated, MbMt-hypermethylated, or MbMt-hypomethylated genes with binding motifs for the myogenic transcription factors MYOG, MYOD1, or the E-protein heterodimer E12/E47 (432, 740 and 528 sites, respectively) attests to the muscle-relevance of the myogenic DM sites (Table S2A).
For MbMt-hypomethylated sites, muscle-protein terms were not among the most significantly overrepresented GO categories (Table S2A) even though they were for genes displaying Mt overexpression.Citation13 Instead, small GTPase regulator activity (660 sites) and other signaling terms as well as focal adhesion were the most strongly associated with hypomethylation. Muscle-hypomethylated sites exhibited the strongest associations with several GO terms related to muscle constituents, namely, contractile fiber (475 sites) and actin cytoskeleton (856 sites).
To look for associations between myogenic differential methylation and expression, we mined data from our previous exon-based expression microarray study of Mb or Mt samples vs. 19 types of non-muscle cell cultures.Citation13 Despite often strong upregulation of expression at the Mb or Mt stages, 9 of 32 contractile-fiber genes (including OBSCN, see below) were heavily methylated in Mb or Mt at sites that displayed hypomethylation in muscle (Table S3). This suggests that DNA hypomethylation was often a consequence of the initiation of expression rather than its precedent, although it might thereby reinforce expression. However, this was not always the case, e.g., the sarcoplasmic protein-encoding CASQ1 gene exhibited little expression in Mb and much more in Mt (Table S3) but had an intragenic site that was hypomethylated in all Mb as well as Mt samples.
For many homeobox genes with MbMt hypermethylation, there was the expected inverse correlation between the hypermethylation and expression in Mb and Mt determined by expression microarray profiling. Of 68 MbMt-hypermethylated homeobox genes represented on the expression microarray, 30 were significantly downregulated, 7 significantly upregulated, and 31 displayed no significant difference in expression in myogenic vs. non-myogenic cells, often with low levels of RNA in most cell types (data not shown). There was enrichment within 5 kb of the TSS of MbMt-hypermethylated sites in homeobox genes, which was greater than that for muscle-hypomethylated sites in contractile fiber genes ( and , green bars). These results suggest that myogenic hypermethylation is helping to downregulate expression of many of the homeobox genes from the extended promoter region in myogenic vs. non-myogenic cells while for others it may be just reinforcing repressive chromatin marks.
Three of the six T-box genes with MbMt hypermethylation are members of the TBX1 subfamily of TBX genes.Citation26 These three genes were strongly upregulated in myogenic progenitor cells despite having hypermethylated sites within 3 kb upstream (TBX15, TBX18 and TBX1). Two of them (TBX15 and TBX1) had > 20 hypermethylated sites in the first exon or intron (Table S3). TBX1 exhibited the second highest number of MbMt- hypermethylated sites per gene (131), and SIM2, the highest number (178). SIM2 is a candidate gene for Down syndrome and, like TBX1, encodes a master regulator transcription factor whose developmental roles include those in skeletal muscle.Citation27 Like TBX1, SIM2 had many MbMt-hypermethylated sites in the extended promoter region and specific expression in Mb samples (data not shown). Moreover, no overall relationship was seen between myogenesis-specific expression and myogenic hyper- or hypo-methylation when all genes with more than 1 MbMt DM site were considered (). Whether muscle-hypomethylated genes with more than 1 hypomethylated site or those with at least 10 were considered, only about 25% of these genes were associated with a muscle functional term (muscle phenotype, myopathy, or at least the 90th percentile expression in muscle; www.genecards.org, GeneDecks, data not shown). Therefore, only certain genes or small subgroups of genes, like homeobox genes in Mb and Mt, displayed such associations.
Table 1. Most of the myogenesis-associated differential methylation does not display a simple correlation with expression levels of the closest genea
DNA methylation in myotubes vs. myoblasts and FSHD vs. control myogenic cells
Comparison of Mb and Mt samples to each other (three each, excluding the FSHD samples) indicated less than 5% as much differential methylation between these two myogenic progenitor cell types as for comparisons of myogenic cells or tissues with non-myogenic samples. In this part of the study, we considered only autosomal DM sites because the set of Mb vs. Mt DM sites was enriched in X-linked sites (12% of all DM sites vs. 2% for all other classes of DM sites), probably as an artifact of the small sample size and different genders in the Mb vs. Mt comparison. Only 583 autosomal sites exhibited significant differential methylation in Mt vs.Mb compared with 19460 for MbMt vs. other cell cultures and 11881 for muscle vs. other tissues (Table S2). Moreover, only 11% of the genes with Mt vs. Mb differential methylation had at least two hypo- or two hyper-methylated DM sites compared with 63 and 59% of genes with differential methylation for MbMt vs. other cell cultures or muscle vs. other tissues, respectively (). If Mt samples were displaying some of the strong trend toward the loss of myogenic hypermethylated sites seen in muscle vs. Mb-plus-Mt, the ratio of hypo- to hyper-methylated sites for Mt vs. -Mb should have been intermediate to that for the other two comparisons (1.05 and 14.8, respectively; ); but, instead, it was smaller than both (0.4). These results indicate that most of the relatively small number of sites with Mt-vs.-Mb differential methylation (3% the number of MbMt vs. non-muscle cell DM sites) is due to sample variation. Although the small sample size (three Mb vs. three Mt samples) may have limited the sensitivity of the comparison to detect infrequent Mt vs. Mb differential methylation, it is clear that there was much more extensive differential methylation between Mt or Mb muscle progenitor cells and skeletal muscle than between the Mb and Mt stages ().
Because FSHD Mb and Mt samples were shown to have highly significant dysregulation of expression of some of their genes relative to controls,Citation13,Citation28 we compared their DNA methylation profiles (four FSHD to five normal controls and one disease-control; ). There were 1348 autosomal DM sites, which was only 7−11% of the DM sites for the comparisons of myogenic progenitors to non-muscle cells or muscle to non-muscle tissues (Table S2). That the number of hypo- vs. hyper-methylated sites was so skewed (84% of these FSHD vs. control DM sites was hypomethylated) suggests biological relevance. The percentage of genes associated with at least two hypo- or two hyper-methylated sites in FSHD vs. control myogenic progenitors was 24%, twice as much as for the Mb vs. Mt comparison but much less than for myogenic-non-myogenic comparisons (). There was significant enrichment for homeobox genes but this enrichment was small compared with myogenic-non-myogenic comparisons (Table S2). However, as described below, an independent test of two FSHD-hypomethylated sites associated with a homeobox gene did not show significant FSHD differential methylation.
Total genomic 5-hmC in muscle vs. myogenic progenitors
Upon mining data from our previous expression microarray profiling,Citation13 we found significant increases in steady-state levels of RNA encoding the 5-hmC-generating TET1 and TET2 enzymes in control Mb or Mt vs. the 19 other cell types (p = 3 × 10−5 to < 1 × 10−6) but not for TET3. TET1 and TET2 RNA levels in these myogenic progenitor cells were higher than those of all the other nontransformed cell types analyzed, with the exception of H1 embryonal stem cells (ESC), which had very high levels of TET1 RNA, as expected.Citation4 The analyzed non-transformed cell types were skin fibroblasts, astrocytes, hepatocytes, human umbilical cord endothelial cells (HUVEC), human mammary epithelial cells (HMEC), melanocytes, normal human epidermal keratinocytes (NHEK), and osteoblasts in addition to Mb, Mt, and ESC. The ratio of the average TET1 RNA signals for Mb vs. the average signal from the nontransformed, non-ESC cell types studied was 6.5, and for Mt, 2.8. The respective increases for TET2 were 3.6 and 3.4.
Given the evidence that 5-hmC residues in DNA can function as an intermediate in the conversion of 5-mC to unmethylated C residues,Citation29 we analyzed relative levels of genomic 5-hmC in an enzymatic assay for the transfer of glucose to 5-hmC, with nonglucosylated phage T4 DNA as a standard.Citation16 Skeletal muscle DNA had an average 5-hmC level of 0.077 mol%, which was about twice that of control or FSHD myoblasts or myotubes (p < 10−4; Table S4). The overall genomic 5-hmC contents of the Mb, Mt, skin fibroblast, and sperm samples and the amniocytes and chorionic villus-derived cells at passage 2 to 3 were similar (0.030 to 0.043 mol%). As expected,Citation5,Citation16 cerebellum DNA had very much higher 5-hmC levels than the other samples (0.57 mol%). The decrease in 5-hmC levels in DNA from leukocytes to an Epstein-Barr virus-transformed B-cell line (lymphoblastoid cell line, LCL) could be due to the effects of in vitro oncogenic transformation.Citation15
Myogenic hypermethylation profiles in genes expressed in myoblasts: PAX3 and TBX1.
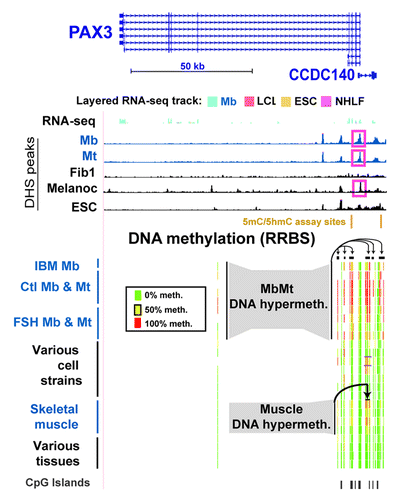
Like many other homeobox or T-box transcription factor-encoding genes, PAX3 andTBX1, were hypermethylated in Mb and Mt. Their DNA and chromatin epigenetics were examined in detail to gain insight into the probable functional significance of myogenesis-associated epigenetic marks. These two genes have important roles in muscle or limb formation and other types of organogenesis and generate alternatively spliced RNAs encoding different protein isoforms.Citation26,Citation30,Citation31 In addition to the 28 MbMt-hypermethylated sites in the gene body of PAX3, there were 58 MbMt-hypermethylated sites in the divergently transcribed, partly overlapping CCDC140 gene or CCDC140’s downstream region. Both genes were weakly, but selectively, transcribed in Mb (Figs. Two and S2a). The myogenic hypermethylated sites within CCDC140 were mostly in DNA sequences conserved between rodents and humans, although this gene of unknown function is primate-specific. The CCDC140-associated sites with MbMt hypermethylation were 1.8−9.2 kb upstream to the PAX3 TSS. Nine MbMt-hypermethylated sites within the single intron of CCDC140 were retained in muscle. These results suggest that, in the muscle lineage, CCDC140 intragenic sequences act in cis as PAX3 transcription regulatory elements, in addition to any independent gene functionality that they might have. Consistent with this interpretation, there are neural-specific and hypaxial somite enhancers within the mouse region similar to the single intron of CCDC140 and immediately downstream of the CCDC140-like region in addition to a second neural enhancer in intron 4 of Pax3.Citation31,Citation32,Citation33 Throughout the PAX3/CCDC140 region, there was a significant, but low, enrichment in H3K9me3 in Mb and Mt (Fig. S2A). At the bidirectional promoter region there were clusters of MbMt-hypermethylated sites on either side of an unmethylated CGI and a DNaseI hypersensitive site (DHS) that was specific for Mb, Mt, and melanocytes (, pink boxes). We used the Cufflinks tool for transcript analysis of RNA-seq dataCitation34 to quantify RNA-seq-determined expression levels (ENCODE/CalTech)Citation35 of different RNA isoforms. We found that there were low concentrations of various PAX3 RNA isoforms (Table S5), including several described in human skeletal muscle.Citation36 Consistent with PAX3’s role in adult melanocyte stem cell differentiation,Citation18 it displayed DM sites, namely, a cluster of CpG sites with DNA methylation preferentially in melanocytes that was downstream of CCDC140 (Fig. S2b). This region partially overlapped sites enriched in methylation in Mb.
Figure 2. Myogenic hypermethylation at PAX3 and the adjacent CCDC140 gene region. From the UCSC Genome Browser (http://genome.ucsc.edu), the following tracks are shown for the PAX3 and CCDC140 gene region (chr2:223,058,677–223,174,523; all coordinates are relative to hg19): RefSeq genes, RNA-seq data (not strand-specific; CalTech); DNase-seq data (this study); and representative RRBS data (this study). The cell strains and tissues illustrated are as follows: IBM Mb, Ctl Mb3, Ctl Mt3, Ctl Mb7, Ctl Mt7, FSH Mb5, FSH Mt5, FSH Mb8, FSH Mt8, LCL, Fib1, Fib2, Fib3, HMEC, SAEC, ESC, Muscle A1, Muscle A2, Muscle B1, Muscle B2, leukocyte, skin, brain and lung. The short horizontal black bars for the RRBS tracks indicate clusters of CpG hypermethylation in Mb, Mt, muscle, and Fib2 and Fib3.
TBX1, the other transcription factor gene examined in detail, had five times more hypermethylated sites in the MbMt set (131) than in skeletal muscle (26; ; Fig. S3A). It was upregulated in both progenitor cells (Table S5) and skeletal muscle tissue.Citation37,Citation38 Cortese et al.Citation39 had previously studied methylation just in the promoter region at the tissue level and found hypermethylation in skeletal muscle vs. heart muscle or liver. Consistent with preferential expression of TBX1 in Mb and Mt (), these myogenic progenitor cells had a broad band of H3K36me3 enrichment (Fig. S3B) not seen in the other examined cell types except, to a lesser extent, in HUVEC, which also expressed this gene (Table S5). H3K36me3 is typically found downstream of the 5′ end of active genes.Citation40 Remarkably, a gap in this band of H3K36me3 signal was centered over a cluster of 79 MbMt-hypermethylated sites (; Fig. S3B) overlapping a CGI and exons 7 - 9.
Figure 3. Predominant myogenic hypermethylation at TBX1, including the promoter region, despite high expression. TBX1 (chr22:19,732,116–19,771,300) epigenetic tracks from the UCSC Genome Browser are illustrated as in with the additions of tracks for sites exhibiting significant myogenic hypo- or hypermethylation (this study); histone H3 modifications (Broad); and CTCF ChIP-seq profiles (Broad). A cluster of MbMt-hypermethylated sites is shown in an expanded view (chr22:19,744,510–19,744,710) at the bottom of the figure with the following representative cell types: Ctl Mb3, Ctl Mt3, Ctl Mb7, Ctl Mt7, LCL, Fib1, osteoblasts, HMEC, IMR-90, small airway epithelial cells (SA epith), and ESC for sequencing reads of ≥ 5.
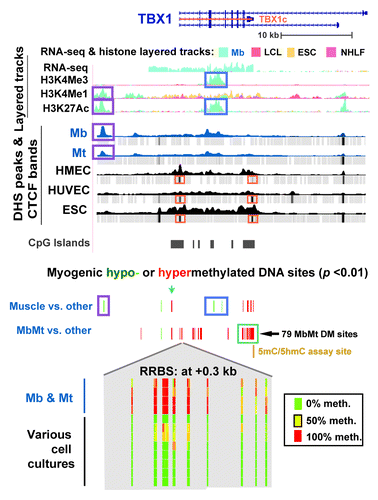
Besides the large clusters of MbMt-hypermethylated DM sites in the interior of TBX1, there were hypermethylated sites in intron 1, including a triplet of MbMt- and muscle-hypermethylated sites only ~100 bp downstream of the TSS (). Surprisingly, in the extended promoter region, there was no peak of H3K4 methylation in Mb, Mt, or HUVEC nor was there a DHS, as typically seen in active or poised promoters.Citation41 Nonetheless, analysis of RNA-seq profiles (ENCODE/CalTech) indicated a predominant, full-length canonical transcript in Mb and HUVEC (2082 nt; NM_080647, TBX1c) starting at the single canonical promoter region and ending at the cluster of 79 MbMt-hypermethylated sites (Table S5). Instead of the conventional peak of H3K4me3 at an active promoter region there was only a broad and strong H3K4me3 peak centered over intron 3 in Mb, Mt, and HUVEC, the only expressing cell types. It was surrounded on either side by MbMt-hypermethylated sites. In muscle, this peak was significantly hypomethylated at CpGs vs. non-muscle tissues (, blue box). In Mb and Mt, this same region was mostly unmethylated even though the CpG sites did not reach the level of significant differences from the non-muscle cell cultures. From this region, two minor transcripts seemed to originate that had retained intronic sequences (Table S5). Examination of small transcript (20−200 nt) RNA-seq data (ENCODE/Cold Spring Harbor) did not indicate any small intragenic ncRNA originating in this internal promoter-like chromatin.
In the canonical, 5′ promoter region of TBX1, there was a cluster of MbMt- and muscle-hypermethylated sites at -1.2 kb (, blue arrow). At -2.6 kb, there was a single MbMt- and muscle-hypomethylated site () that is near a subregion of Mb-specific AS transcription (Fig. S3A). The only non-muscle cell culture that was mostly unmethylated at this -2.6 kb site was choroid plexus epithelial cells, a finding that might be relevant to the proposed neurological functioning of TBX1.Citation42 Yet further upstream at -11 kb, there was a muscle- and MbMt-hypomethylated CpG site near a Mb- and Mt-specific DHS and myogenic enhancer-like peaks of H3K4Me1 and H3K27Ac (, purple boxes). Another type of epigenetic mark that distinguished Mb and Mt was the differential presence of a binding site for the DNA binding protein CTCF, which helps organize chromatin in cis.Citation43 From CTCF ChIP-seq (ENCODE/Broad), only Mb and Mt lacked CTCF binding sites in the TSS region and at the 3′ end of the TBX1c gene isoform (; Fig. S3B). Therefore, the MbMt-hypermethylated subregions of TBX1 at the beginning and end of theTBX1c isoform indicated in might be linked to alterations in the gene’s higher-order structure via CTCF. As expected from the high level of expression of this gene in myogenic cells, including skeletal muscle tissue, Mb and Mt did not have the broad band of repression-associated H3K27Me3 and EZH2 bindingCitation44 across the upstream and 5′ end of the gene that was seen in non-myogenic cells (Fig. S3B).
Assay for 5-hmC vs. 5-mC at specific sites in TBX1 and PAX3/CCDC140 regions
We used an enzymatic assay to quantify 5-hmC and 5-mC at CCGG sites (Epimark, New England Biolabs) overlapping three MbMt-hypermethylated CGs associated with PAX3 or TBX1. Like the above-described enzymatic assay for total genomic 5-hmC, this site-specific assay involves incubation with T4 β-glucosyltransferase. After incubation or a mock-enzyme incubation, aliquots are digested with MspI, which can cleave CCGG sites whether or not they are methylated or hydroxymethylated at the internal C residue but not if they contain glucosylated 5-hmC. In parallel, digestions are done with HpaII, which can cleave CCGG sites only if they are unmodified at the internal C, and other aliquots are used as uncut controls. Upon real-time PCR of all of these mixtures, 5-hmC and 5-mC in the middle of the analyzed MspI site is quantified by subtraction of the threshold cycles (Ct).
The first MspI site that we examined was 7.9 kb upstream of PAX3 in a CGI shore (0.3 kb from the CGI) and 1.6 kb downstream of CCDC140, the immediate neighbor of PAX3. The second site was in exon 9 of TBX1 in the CGI with 79 MbMt-hypermethylated sites (8% total CpG; and ). These genes are weakly (PAX3 and CCDC140) or strongly (TBX1) transcribed in Mb with preferential transcription in myogenic vs. non-myogenic cells. We tested four to six biological replicates of normal-control Mb, FSHD Mb, normal skeletal muscle, heart, leukocyte and cerebellum, almost all of which had not been included in the RRBS analysis. We found that essentially all the modification of C residues for both sites was 5-mC, rather than 5-hmC, in Mb samples (). For the PAX3-upstream, CCDC140-downstream site, the 5-mC status is consistent with the cell type-specific enrichment of H3K9me3 signal throughout PAX3/CCDC140 region in Mb (Fig. S2A). Unlike the Mb samples, the skeletal muscle samples were enriched in 5-hmC at both the TBX1 and PAX3 sites relative to all other samples, including cerebellum (T-test, p < 0.001). Therefore, these genomic regions reflect the higher global 5-hmC content in skeletal muscle vs. Mb or Mt, as described above. Remarkably, in a given sample, the ratios of 5-hmC to 5-mC were similar for the tested PAX3 upstream site and the TBX1 exon 9 site even though these ratios differed much among the skeletal muscle samples. At a MbMt-hypermethylated site in intron 3 of PAX3, similar results for Mb and skeletal muscle vs. non-muscle samples were found in a smaller set of samples (2 each; data not shown).
Figure 4. Only skeletal muscle had considerable 5-hmC at assayed CpG sites associated with TBX1 or CCDC140/PAX3. Each of the indicated samples was assayed with or without incubation with T4 β-glucosyltransferase or mock treatment, followed by MspI, HpaII or mock incubation, and then quantitative PCR to determine 5-hmC and 5-mC levels at the assayed sites.
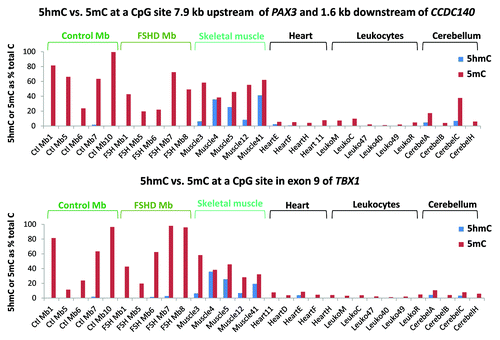
The above enzyme assays also confirmed the RRBS findings of hypermethylation in the muscle lineage. We chose the PAX3-upstream site for testing because, according to the RRBS data, it was not only a MbMt-hypermethylated site but also one of the 17 sites in this region that was significantly hypomethylated (most sites at p < 10−6) in FSHD MbMt vs. analogous control MbMt (). The above-mentioned PAX3 intronic site also displayed FSHD hypomethylation in RRBS data set. However, the results from the enzyme-PCR assay at the PAX3-upstream site did not give statistically significant differences at the p < 0.05 level (t-test, p = 0.07) between FSHD and control Mb samples (). Similarly, the site in PAX3 intron 3 that was examined in three FSHD and two normal-control Mb samples did not validate the RRBS-indicated difference between FSHD samples and controls (data not shown). Therefore, our finding of a modest number of RRBS sites with significant FSHD vs. control differential methylation by RRBS might just be due to individual variation and small sample sizes in the FSHD vs. control comparison. This would be consistent with the FSHD DM sites lacking strong associations with functional groups other than homeobox genes and lacking clustering of most sites unlike MbMt vs. non-muscle DM sites and muscle vs. non-muscle DM sites (; Table S2A).
Myogenic hypomethylation profiles: MYH7B/MIR499 and OBSCN.
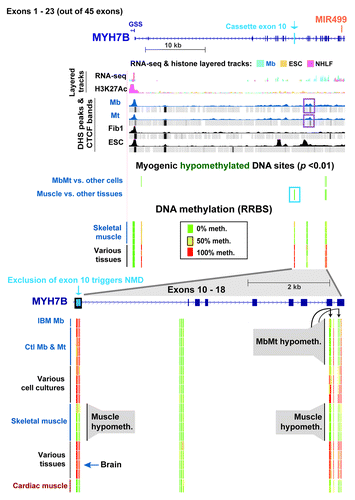
To elucidate the functionality of gene-body hypomethylation in the muscle lineage, the DNA and chromatin epigenetics of OBSCN (> 80 exons) and MYH7B (45 exons) were examined. These genes had 72 and 20 intragenic muscle-hypomethylated CpG sites and 2 and 12 MbMt-hypomethylated CpGs, respectively (Figs. 5 and 6). Both encode proteins that are found mostly in skeletal muscle and heart and, at lower levels, in parts of the brain and certain other tissues.Citation23,Citation37,Citation38,Citation45,Citation46 The OBSCN protein product is involved in sarcomere structure and important in the skeletal and heart muscle lineages.Citation23 In contrast, MYH7B is important in skeletal and cardiac muscle mainly because of its microRNA, MIR499, which is released from an intron (human intron 22), rather than because of its protein product.Citation21,Citation47 Although we did not include any muscle samples other than skeletal muscle in our statistical analyses of myogenesis-associated DM sites, we also used heart DNA for RRBS profiling of DNA methylation. Many of the MYH7B sites that displayed tissue-associated hypomethylation in skeletal muscle also had little or no methylation in heart DNA; however, in OBSCN there was less sharing of hypomethylated sites between these two types of muscle ( and ; Figs S4 and S5). In addition, for OBSCN, MYH7B, PAX3 and TBX1, MbMt DM sites usually showed a similar methylation status in IBM Mb as in the normal-control Mb samples (, and ; Fig. S3A).
Figure 5. Myogenic hypomethylation in the OBSCN, which encodes a giant muscle-associated protein. DNA and chromatin epigenetic marks for OBSCN (chr1:228,378,622–228,575,138; including C1orf145) are indicated as in the previous figures with the addition of Mt and Mb data for histone H3 methylation and acetylation (Broad, http://genome.ucsc.edu) shown in bar format. At the bottom of the figure is an enlarged view of skeletal muscle-associated hypomethylation at the exon 8/intron 8 border (chr1:228,404,946–228,405,003) for the following samples: IBM Mb, Ctl Mb3, Ctl Mt3, Ctl Mb7, Ctl Mt7,Fib1, HMEC, ESC, Muscle A1, Muscle A2, Muscle B1, Muscle B2, leukocyte, kidney, stomach and testis. Cardiac muscle, which was not included in the set of samples for determination of muscle-specific DM sites, is also shown. Myogenesis-associated epigenetic marks that are referenced in the text are indicated by boxes or triangles.
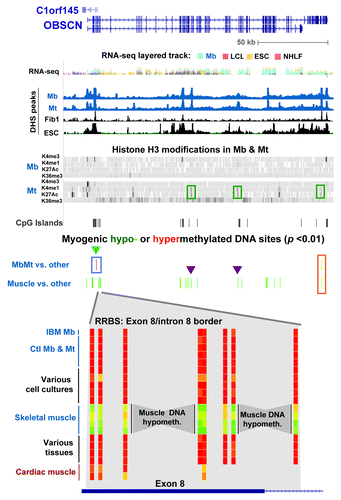
Figure 6. Myogenesis hypomethylated sites in MYH7B/MIR499 over an exon whose inclusion triggers nonsense-mediated decay of mRNA. DNA and chromatin epigenetic marks are indicated as in the previous figures for half of the exons of MYH7B (exons 1–23) and the 5′ end of GSS (chr20:33,542,729–33,578,756). An expanded view of exons 10–18 (chr20:33,570,058–33,575,875) is shown at the bottom of the figure. The representative cell strains and tissues illustrated are as follows: IBM Mb, Ctl Mb3, Ctl Mt3, Ctl Mb7, Ctl Mt7, LCL, Fib1, HMEC, ESC, Muscle A1, Muscle A2, Muscle B1, Muscle B2, leukocyte, kidney, brain, stomach and cardiac muscle. DHS and CTCF peaks that were specific to Mb and Mt are boxed in purple.
The many muscle-hypomethylated sites in OBSCN and MYH7B were mostly in internal exons and introns. The ratios of DM sites in internal exons vs. introns were 2.5 and 3.0 for OBSCN and MYH7B, respectively. This is considerably higher than the respective ratios of 1.0 and 1.6 for all RRBS-detected CpGs in these two genes and 0.36 for all RRBS-detected CpGs in all genes associated with muscle-lineage DM sites (Table S2C). These results suggest that the generation of alternate RNA isoforms for both genesCitation21,Citation23,Citation48 might be controlled, in part, by differential DNA methylation. NEB and TTN, two other genes that encode giant sarcomeric proteins,Citation23 also displayed skeletal muscle-associated DNA hypomethylation preferentially in internal exons (5 and 8 myogenic DM sites, respectively; Table S3).
The only two OBSCN MbMt-hypomethylated sites were 13 bp apart at the 3′ end of the gene (, orange box). Their retention in skeletal muscle signifies their probable importance. These sites and two of the small intragenic clusters of muscle-hypomethylated sites (, purple triangles) resided in Mt chromatin that was enhancer-like in its enrichment, albeit low, in H3K4me1 and H3K27ac. The increase in these enhancer-like H3 modifications in Mt relative to Mb paralleled the much higher, but still modest, level of expression of OBSCN in Mt vs. Mb as determined from our expression microarray profiling. Many of the DM sites in this gene overlapped exons or were near putative alternative TSS (UCSC genes track in Fig. S4). At the Mb stage, an assortment of variant RNA isoforms were seen, but not the major muscle-associated OBSCN isoform a or b transcripts (Table S5).
Muscle-lineage DNA hypermethylation was observed at only two CpG sites, 11 bp apart, and only in Mb and Mt. These were located at the border of an OBSCN CGI overlapping exons 2 – 4 (, blue triangle). These sites were immediately upstream of the 5′ end of novel antisense transcripts (C1orf145 and AK056556), whose 3′ ends are in OBSCN’s promoter region (Fig. S4). The MbMt-hypermethylated sites were surrounded by clusters of muscle-hypomethylated sites (, bottom) that were indicative of DNA demethylation occurring after the Mt stage. OBSCN protein is implicated in the assembly and stabilization myofibrilsCitation23 and is most abundant in the muscle lineage, especially in skeletal muscle tissue.Citation37,Citation38 When hypomethylated in muscle, this subregion of the OBSCN gene might upregulate expression of the antisense transcripts and thereby help increase expression of OBSCN after formation of myofibrils, which form subsequent to the Mt stage.
MYH7B displayed remarkably specific skeletal and cardiac muscle hypomethylation in exon 10 and exons 17–18. These DM sites surround small Mb- and Mt-specific DHS and a CTCF binding site mostly associated with Mb and Mt (, purple boxes; Fig. S5). In skeletal muscle (and heart) there was an apparent spreading of MbMt-hypomethylation in exons 17 and 18 to the intron between them and to an additional site in exon 18. Importantly, exon 10 had seven CpGs that were hypomethylated in skeletal muscle (and heart) but highly methylated in all Mb and Mt samples and all non-muscle tissue samples. This unusual exon is largely excluded from the mature mRNA in C2C12 Mb and Mt, cardiac muscle, and many types of skeletal muscle but it is often included in brain.Citation21 Its exclusion leads to the creation of a premature nonsense codon and nonsense-mediated decay (NMD) of the mRNA in the cytoplasm.Citation21 We found that MYH7B RNA was present at too low steady-state levels in Mb for accurate analysis of RNA isoforms from RNA-seq data. This probably reflects much NMD of the transcript in human Mb and Mt, as previously observed for C2C12 cells. Nonetheless, there were increases in steady-state RNA levels for this gene in human (our microarray expression data) and C2C12Citation21 Mt vs. Mb. Our results suggest that differential methylation of exon 10 helps guide alternative splicing of this gene.
Discussion
Our RRBS profiling of DNA methylation in Mb, Mt, skeletal muscle, and non-muscle samples indicates that myogenesis-associated changes in DNA methylation before the Mb stage involve similar amounts of demethylation and de novo methylation but, after the Mt stage, predominantly demethylation. Skeletal muscle lost > 90% of the MbMt-hypermethylated sites and acquired many additional hypomethylated sites. Much of this loss of hypermethylation was in genes encoding sequence-specific transcription factors, especially homeobox and T-box genes. There was highly significant enrichment in muscle-associated genes among muscle-hypomethylated genes. Nonetheless, only about 25% of the genes associated with muscle hypomethylation were specifically expressed in muscle or linked to a muscle phenotype. In addition, there were no simple overall relationships between MbMt-hypo- or hypermethylation and Mb- or Mt-associated expression, only for certain subsets of these genes, like contractile fiber genes.
Because the majority of nuclei in skeletal muscle tissue samples are postmitotic, the decreased DNA methylation in skeletal muscle vs. Mb and Mt cannot be due to passive demethylation, which relies on replicative DNA synthesis.Citation49 Instead, our results offer the first evidence there is active, genome-wide demethylation at CpGs in skeletal muscle. Implicated in pathways for active demethylation of mammalian DNA are TET1, TET2, and TET3 enzymes. They catalyze the formation of 5-hmC as well as 5-formylcytosine (5-fC) and 5-carboxymethylcytosine (5-caC) from 5-hmC.Citation5,Citation50,Citation51 Evidence for several DNA-demethylating pathways downstream of the TET-catalyzed formation of 5-hmC, 5-fC, or 5-caC has been reported although the details remain to be definitively established.Citation49,Citation52-Citation54 Surprisingly, we found that Mb and Mt samples have much higher levels of TET2 RNA than any of ten analyzed non-muscle cell strains and more TET1 RNA, with the expected exceptionCitation4 of embryonal stem cells. Skeletal muscle also has a high level of TET1 RNA relative to many tissue types,Citation55 and TET2 was expressed in only about half of the tested tissue types, including skeletal muscle.Citation56 TET-catalyzed hydroxymethylation of 5-mC can also lead to passive DNA demethylation by interfering specifically with DNMT1 maintenance methylation.Citation57 TET1- or TET2-faciliated passive demethylation might occur at a pre-myoblast, cell cycling stage to help generate the Mb- and Mt-associated DNA hypomethylation that we observed.
Using an enzymatic assay for global genomic 5-hmC, we showed that overall levels of this 5-mC-derived base were twice as high in skeletal muscle as in Mb or Mt samples but similar in Mb, Mt, cultured skin fibroblasts, and early-passage chorionic villus cells and amniocytes. These results indicate that some of the hydroxylation of genomic 5-mC catalyzed by TET enzymes in the muscle lineage leads to increases in stably maintained 5-hmC DNA residues after the Mt stage. With another enzymatic assay on various samples, we demonstrated that considerable amounts of 5-hmC were detected only in skeletal muscle tissue at three tested CpG sites in upstream, intronic, or 3′-terminal clusters of CpGs exhibiting MbMt hypermethylation. In contrast, Mb had 5-mC as essentially the only modified C residue at these MbMt-hypermethylated sites. The genes tested were PAX3 andTBX1, homeobox and T-box genes, respectively. They are preferentially expressed in myogenic progenitor cells, have important roles in myogenesis and other developmental programs,Citation17,Citation19,Citation30 and, like many other homeobox and T-box transcription factor genes, were enriched in MbMt-hypermethylated sites. At the tissue level, skeletal muscle has elevated levels of TBX1 RNA and of one PAX3 RNA isoformCitation36,Citation37,Citation58 vs. most non-muscle tissues and retained some of the MbMt-hypermethylation ( and ).
We propose that myogenesis-associated 5-mC and 5-hmC CpG sites help positively or negatively regulate transcription or co-transcriptional posttranscriptional RNA processing depending on the sequence and developmental context. Evidence for 5-hmC as well as 5-mC performing such roles has recently been reported for non-myogenic DM sites.Citation3,Citation49,Citation59-Citation62 Cell type-specific epigenetic marks, including enhancer-type chromatin modifications, are often inside the body of genes.Citation63,Citation64 We found that about 35−45% of all RRBS-detected MbMt- or muscle-DM sites were in internal exons or introns. One of the four genes that we highlighted was OBSCN, which encodes a giant structural and signaling protein associated with the formation of myofibrils subsequent to the Mt stage.Citation23 It had 72 intragenic muscle-hypomethylated sites distributed over many of its > 80 internal exons and introns. Most of the OBSCN exons encode modular protein domains with compatible splice donor and acceptor sites enabling complex splicing patterns to produce functional protein isoforms. Therefore, modulation of alternative splicing by differential gene methylation could lead to differentiation-related differences in the resulting proteins. This is consistent with Mb having OBSCN RNA isoforms that differed from those associated with skeletal muscle and Mb lacking most of the OBSCN DNA hypomethylation seen in skeletal muscle. At two intragenic OBSCN subregions that displayed muscle- but not MbMt-hypomethylation, Mt samples had chromatin epigenetic features that were typical of weak enhancers. This indicates enhancer-like chromatin epigenetic changes preceding post-mitotic DNA demethylation for this gene.
We found evidence for cis-regulation of transcription of one gene by a differentially methylated internal regulatory element of a neighboring gene. PAX3 encodes an essential homeobox/paired-box protein that contributes to early striated muscle development and partly overlaps 5′-to-5′ the CCDC140 gene. CCDC140 is a primate-specific gene that encodes a protein of unknown function. Mb, Mt, and skeletal muscle had clusters of myogenic hypermethylation (including 5-hmC in skeletal muscle) in intron 3 of PAX3 and also in the single CCDC140 intron and downstream of CCDC140 (). The myogenic DM sites in the CCDC140 intron and downstream of the gene are ~1−1.5 kb from neural or hypaxial somite enhancers of mouse Pax3.Citation31,Citation32 They are located in sequences that are highly conserved between mice and humans despite the lack of a CCDC140 gene in mice. Such conservation is also seen in the region of MbMt hypermethylation in PAX3 intron 3. These MbMt-hypermethylated sites and the less abundant muscle-hypermethylated sites in CCDC140 and PAX3 introns might prevent leaky activation of development-specific non-muscle enhancers of PAX3 in the muscle lineage.
TBX1 offers an especially intriguing example of myogenic differential methylation in a gene of major clinical importance. Haploinsufficiency of TBX1 is responsible for most of the symptoms of the DiGeorge/velo-cardio-facial syndrome, the most common microdeletion syndrome in humans.Citation65,Citation66 Symptoms include congenital heart and craniofacial abnormalities, skeletal muscle hypotonia, and increased risk of psychiatric disorders. Variations in expression levels in vivo may be relevant to the disparities in genotype-phenotype correlations.Citation65 In addition to its role in the above syndrome, TBX1 has been separately implicated in behavioral disorders.Citation42 Control of transcription of this gene and posttranscriptional regulationCitation67 have to be exquisitely regulated because both haploinsufficiency and overexpression are deleterious, and this gene is differentially expressed in different cell types and tissues. TBX1 had an extraordinarily high number (127) of intragenic MbMt-hypermethylated sites as well as one MbMt- and muscle-hypomethylated site and three MbMt- and muscle-hypermethylated sites in the 3-kb upstream region. From exon 7 to exon 9 of the TBX1 isoform c, there is a 1.7-kb CGI that contains 79 of the MbMt hypermethylated sites and a moderate CpG content (8%). Moderate CpG contents in CGIs favor multiple 5-hmC residues,Citation68 and indeed, skeletal muscle, though not Mb or other tested tissue types, had considerable 5-hmC at the assayed site in this region ().
There are several indications of the possible functional significance of the dense myogenic hypermethylation of TBX1’s large intragenic CGI and its relationship to preferential expression of TBX1 in myogenic cells. This gene’s 5-mC or 5-hmC residues in the muscle lineage might influence retention of the overlapping cassette exon, which is the terminal exon of TBX1c RNA, the predominant RNA isoform in Mb (Table S5). In Mb and Mt, this CGI has a distinct chromatin structure lacking the H3K36me3 signal of the surrounding sequences which, along with DNA methylation, might help regulate whether exon 9 is included in the mRNA or not.Citation69 This CGI also overlaps chromatin with enhancer- and promoter-type histone modifications in HMEC but not in Mb and Mt samples (Fig. S3). Therefore, its hypermethylation in myogenic cells might downmodulate the activity of an enhancer specific for certain other cell types. Lastly, centered over intron 3 at ~2 kb upstream of this CGI, there was a large peak of H3K4me3 specific to Mb, Mt, and HUVEC as well as hypomethylated CpG sites in muscle (). Surprisingly, these cells contained no other H3K4me3 peak in the TBX1 vicinity even though H3K4me3 is generally associated with active promoters, and this gene was very highly expressed in myogenic progenitor cells and moderately expressed in HUVEC to give predominantly a full-length RNA starting at the canonical promoter. Because H3K4me3 peaks sometimes also indicate active enhancers,Citation64 this H3K4me3 peak with the dense clusters of hypermethylation several kb to either side may be a potent tissue-specific enhancer. It could thereby reverse a pharyngeal arch-associated silencer element detected in the largely orthologous murine intron2-to-intron3 region of Tbx1.Citation65 The complexity of the cis-regulatory elements in TBX1Citation65 is consistent with the complexity that we observed at the epigenetic level and is probably necessary to precisely regulate the gene’s expression in a cell type- and developmentally-specific way. TBX1 not only plays a critical role in regulating the number of myocytes in the developing limbCitation26 but its differential regulation also extends to muscle type (quadriceps, soleus, and gastrocnemius; in order of relative expression).Citation70 Therefore, some of the variability seen in 5-hmC contents of different skeletal muscle samples at the tested exon-9 site in the present study might be explained by a variety of types of skeletal muscle in our samples. The exact muscle type was unknown for these autopsy samples. In contrast to TBX1’s importance in both developing and fully formed skeletal muscle tissue, it is critical for proper heart formationCitation71 but has little or no expression in mouse heart.Citation58 Correspondingly, we observed that most of the hypermethylated and hypomethylated CpGs in skeletal muscle within or immediately upstream of TBX1 did not show a similar methylation status in heart.
MbMt- and skeletal muscle-hypomethylation was seen at many sites within the MYH7B/MIR499 gene. Heart shared a low methylation status at most of these sites with skeletal muscle (). MIR499, the miRNA, which is generated after excision from MYH7B intron 22, helps specify slow vs. fast skeletal muscle fiber type and is implicated in myogenesis, cardiac muscle formation, and cardiac stress-response.Citation21,Citation22,Citation47 MIR499 is present at very different steady-state levels in different types of skeletal muscle and in heart; the relative order of expression is ventricle, soleus, gastrocnemius, and quadriceps.Citation21,Citation47 The MYH7B/MIR499 gene is very unusual because although it is most strongly associated with skeletal and cardiac muscle, its predominant RNA isoform in those tissues skips MYH7B exon 10 and thereby generates only the miRNA. The full-length mRNA with the retained exon 10 that can encode the MYH7B protein is produced in very low amounts in these tissues. The lack of protein product from the exon 10-skipped RNA isoform is due to a shift in reading frame and NMD upon exclusion of exon 10, which causes the destruction of the transcript in the cytoplasm without affecting the levels of the intronic-encoded miRNA.Citation21,Citation22,Citation47
The observed hypomethylation in alternative exon 10 of MYH7B in skeletal and cardiac muscle, but not in Mb or Mt, as well as the hypomethylation in exons 17 and 18, might increase the efficiency of skipping of exon10 in a tissue- and developmental-specific manner. Brain, which was fully methylated in these regions, has much more inclusion of cassette exon 10 in its mRNA.Citation21 The increased ratio of MYH7B protein to MIR499 RNA in brain may be linked to MYH7B protein being involved in the regulation of brain synapses.Citation46 The DNA hypomethylation, the myogenesis-associated CTCF binding site, and the small DHS between exons 10 and 17 might contribute to this tissue-specific splicing by affecting the rate of transcription elongation.Citation61 In addition to association with differentially spliced exons, MbMt-specific hypomethylation in MYH7B probably helps control the levels of transcription. For example, 1 kb downstream of the TSS there was muscle-lineage associated DNA hypomethylation.
Therefore, our study suggests that differential methylation of many genes that are important for forming skeletal muscle, like MYH7B/MIR499, TBX1 and OBSCN, is involved in both regulation of expression levels and cell type-specific processing of pre-mRNA. Consistent with this conclusion, differential DNA methylation has previously been implicated in myogenesis from studies of several specific genes and from treating mouse myoblasts with 5-azacytidine, a DNA methylation inhibitor.Citation72-Citation74 Expression of some genes, e.g., MYH7B and TBX1, varies with muscle fiber-type. Therefore, DNA methylation may also help regulate metabolic and structural changes in skeletal muscle in response to physiological alterations, e.g., exercise,Citation75 given the especially dynamic nature of this tissue.
Materials and Methods
Samples
Mb cell strains used for methylation analysis were propagated from muscle biopsy samples of FSHD and control individuals (Table S1).Citation13 Mt samples were obtained from these myoblast cell strains by serum limitation for 5 d. By immunostaining,Citation13 we demonstrated that all batches of myoblasts contained > 90% desmin-positive cells and that myotube preparations consisted of > 70% of the nuclei in multinucleated cells desmin-positive and myosin heavy chain-positive cells. FSHD myoblasts were derived from clinically and molecularly diagnosed patients with disease-associated shortening of the D4Z4 repeat array. The other cell cultures and tissue samples for DNA methylation profiling are also shown in Table S1.
DNA methylation profiling
High-molecular weight DNA was extracted from Mb and Mt samples and used for reduced representation bisulfite sequence (RRBS) for genome-wide DNA methylation profiling. RRBS was done using MspI to digest 1-µg aliquots of DNA before bisulfite treatment and next-generation sequencing on an Illumina platform.Citation12,Citation76 RRBS data are available from the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=292099017&c=chr1&g=wgEncodeHaibMethylRrbs). In addition, for a few genes, DNA methylation profiles from the Infinium Human Methylation 450 platform (http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=292099619&c=chr20&g=wgEncodeHaibMethyl450), which also uses bisulfite-treated DNA, were used to supplement the RRBS profiles to qualitatively evaluate correlations between DNA methylation and gene expression.
Statistical analyses of DNA methylation
All statistical analyses were performed using R version 2.14.Citation77 BED files containing DNA methylation data for each sample from the UCSC genome browser (http://genome.ucsc.edu) were aggregated into matrices with rows included for each site detected in any sample, with one matrix for tissues and another for cell lines. Both matrices included approximately 2 million sites spanning the 24 chromosomes, and these were stored and accessed using the bigmemory package.Citation78 To assess differential methylation in each of our comparisons, a binomial regression model was fit for each site for which read counts and methylation levels were present for a representative number of samples in each group. Where appropriate, random effects terms were included to adjust for systematic variation due to tissue subtypes and/or technical replication using the nlme4 package.Citation79 The significance of the methylation change at each site was assessed using the p-value for the group effect in each comparison. To increase the specificity of our analyses, we restricted our attention to those sites for which a change in methylation percentage of at least 50% was observed at a significance level of 0.01 or below, and these DM sites were output in the BED file format for further bioinformatics analysis.
The program closestBedCitation80 was employed to determine the nearest gene for each DM site using a subset of the contents of the UCSC refFlat table (http://genome.ucsc.edu/cgi-bin/hgTables, 5/01/12) including only one isoform per gene. We gave precedence first to those isoforms chosen for best fit in our study of exon-based microarray gene expressionCitation13 (Affymetrix) of Mb and Mt and then to those isoforms with the RefSeq accession id “NM*” for protein-encoding genes. In the event that closestBed reported multiple genes overlapping a given site, tie-breaking rules were employed to select a single gene on the basis of criteria such as proximity of the 5′ end to the DM site. In other cases, the first gene encountered in genomic order among multiple equidistant genes not overlapping the given DM site was chosen as the tiebreaker. Thus having determined one output gene isoform record for each input DM site, we composed an R script to count the frequency of occurrence of both hyper- and hypomethylated DM sites in each of the following genomic subregions: 2 kb upstream of TSS, a singleton exon, first non-singleton exon and intron, other internal exons and introns, last exon plus 2 kb downstream, and intergenic regions excluding 2 kb upstream of the TSS or downstream of the TTS.
DNaseI hypersensitivity mapping
Genome-wide mapping of DNaseI-hypersensitive sites (DHS) was done on two of the same batches of control myoblast and myotube samples that were used for RRBS (Table S1). The other two Mb and Mt pairs of samples were from two additional myoblast cell strains and were of similarly high quality when checked by immunostaining as described above. For DHS profiling, intact nuclei were treated with DNaseI and the DNaseI-hypersensitive fraction was analyzed by next-generation sequencing as previously described.Citation81 Data from these three biological replicates were combined for the DHS tracks in the UCSC genome browser’s ENCODE data (genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=292099619&c=chr20&g=wgEncodeOpenChromDnase). DHS profiling of the non-myogenic cell cultures had been previously reported.Citation81,Citation82
Histone modification, CTCF binding, and transcription analyses
Data sets and sample information for histone modifications and CTCF binding; non-strand-specific RNA-seq; and strand-specific RNA-seq [Long RNA-seq, > 200 nt poly(A)+ RNA] were from the ENCODE project (http://genomes.ucsc.edu) via the laboratories of Bradley Bernstein (Broad Institute), Barbara Wold (California Institute of Technology), and Tom Gingeras (Cold Spring Harbor), respectively. RNA isoform analysis and quantification was done with the CuffDiff toolCitation34,Citation83 using the above-mentioned ENCODE non-strand-specific RNA-seq database. Mb and Mt samples for ENCODE histone modification and CTCF profiling and Mb for RNA-seq had been commercially obtained, and no immunostaining for determination of percentage contamination with other cell types was described. In addition, we used previously unreported data from our expression profiling of Mb and Mt vs. 19 types of non-muscle cell cultures on microarrays (Affymetrix Exon 1.0 STCitation13).
Determination of 5-hydroxymethylcytosine levels in genomic DNA and at specific CCGG sites
For the determination of the levels of genomic 5-hmC, a T4 β-glucosyltransferase (β-GT; New England Biolabs) glucosylation assay was performed on 2 µg of purified genomic DNA incubated with UDP-[3H]glucose and β-GT and applied to a DE81 membrane with determination of the membrane-bound radioactivity, as previously described.Citation16 All glucosylation reaction values were corrected for non-specific binding of UDP-[3H]glucose to membranes using a DNA-minus reaction. Using a standard curve made from genomic T4-gt DNA (New England BioLabs), the fmol of 5-hmC per microgram of genomic DNA was calculated and then converted to 5-hmC mol% of total genomic bases as previously described.Citation16 For analysis of levels of 5-hmC and 5-mC at a given MspI site, we used another assay involving β-GT (Epimark, New England Biolabs). DNA samples were treated with β-GT or used for mock reactions, followed by digestion with MspI, HpaII, or mock digestion, and then real-time PCR and assessment of methylation status calculated by subtraction of Ct values according to the manufacturer’s instructions. The forward and reverse primers for PCR were as follows: TBX1, 5′-GTGGGATGGAGGGACGATT-3′ and 5′-CCCAAACCTCGCCTGAAC-3′; PAX3 intron 3, 5′-CCTGAGACTCTCGGAGGAAA-3 and 5′-TTTGGTAACCCCAACCGAA-3′; and CCDC140 downstream/PAX3 upstream, 5′-AAGTTGACTGGAGATTGCGGTTA-3′ and 5′-AGAGGTGGTCGCACAGGA-3′.
| List of abbreviations: 5-hmC | = | 5-hydroxymethylcytosine |
| 5-mC | = | 5-methylcytosine |
| Mb | = | myoblasts |
| Mt | = | myotubes |
| RRBS | = | reduced representation bisulfite sequencing |
| CGI | = | CpG island |
| DM | = | differentially methylated |
| IBM | = | inclusion body myositis |
| FSHD | = | facioscapulohumeral muscular dystrophy |
| MbMt | = | the set of all Mb and Mt samples |
| TSS | = | transcription start site |
| DHS | = | DNaseI hypersensitive site |
| TSS | = | transcription start site |
| TTS | = | transcription termination site |
| GREAT | = | genomic regions enrichment of annotations tool |
| LCL | = | lymphoblastoid cell line |
| NHLF | = | human lung fibroblasts |
| ESC | = | embryonal stem cells |
| HMEC | = | human mammary epithelial cells |
| HUVEC | = | human umbilical vein endothelial cells |
| ChIP | = | chromatin immunoprecipitation |
| AS | = | antisense |
| NMD | = | nonsense-mediated decay |
| ncRNA | = | non-coding RNA |
Additional material
Download Zip (2.7 MB)Acknowledgments
We thank the ENCODE Chromatin Group at the Broad Institute for the chromatin data, and the ENCODE Transcription Group at California Institute of Technology (Barbara Wold’s group at CalTech for the RNA-seq data and Tom Gingeras’ group at Cold Spring Harbor Laboratory; http://genome.ucsc.edu) for making their data available prepublication. We are very grateful to many FSHD patients and their unaffected relatives without the disease-associated 4q35.2 allele; to Drs V. Vedanarayanan and Rabi Tawil for the muscle samples used to generate myoblast cell strains; and to Dr Stephen Hauschka for the donation of the myosin heavy chain MF20 antibody and advice on culturing conditions for optimal myotube formation. This research was supported by grants from the National Institutes of Health to M.E. (NS04885), R.M.M. (NHGRI 5U54HG004576), G.E.C. (NHGRI U54HG004563), from the FSHD Global Research Foundation to M.E. and by a COBRE grant (NIGMS P20GM103518).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ehrlich M, Gama-Sosa MA, Huang L-H, Midgett RM, Kuo KC, McCune RA, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 1982; 10:2709 - 21; http://dx.doi.org/10.1093/nar/10.8.2709; PMID: 7079182
- Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends Biochem Sci 2011; 36:381 - 7; PMID: 21497094
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929 - 30; http://dx.doi.org/10.1126/science.1169786; PMID: 19372393
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011; 473:398 - 402; http://dx.doi.org/10.1038/nature10008; PMID: 21460836
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011; 145:423 - 34; http://dx.doi.org/10.1016/j.cell.2011.03.022; PMID: 21496894
- Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res 2011; 39:5015 - 24; http://dx.doi.org/10.1093/nar/gkr120; PMID: 21378125
- Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2010; 2:657 - 69; http://dx.doi.org/10.2217/epi.10.44; PMID: 21339843
- Cotton AM, Lam L, Affleck JG, Wilson IM, Peñaherrera MS, McFadden DE, et al. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum Genet 2011; 130:187 - 201; http://dx.doi.org/10.1007/s00439-011-1007-8; PMID: 21597963
- Yuen RK, Neumann SM, Fok AK, Peñaherrera MS, McFadden DE, Robinson WP, et al. Extensive epigenetic reprogramming in human somatic tissues between fetus and adult. Epigenetics Chromatin 2011; 4:7; http://dx.doi.org/10.1186/1756-8935-4-7; PMID: 21545704
- Isagawa T, Nagae G, Shiraki N, Fujita T, Sato N, Ishikawa S, et al. DNA methylation profiling of embryonic stem cell differentiation into the three germ layers. PLoS One 2011; 6:e26052; http://dx.doi.org/10.1371/journal.pone.0026052; PMID: 22016810
- Berdasco M, Melguizo C, Prados J, Gómez A, Alaminos M, Pujana MA, et al. DNA methylation plasticity of human adipose-derived stem cells in lineage commitment. Am J Pathol 2012; 181:2079 - 93; http://dx.doi.org/10.1016/j.ajpath.2012.08.016; PMID: 23031258
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:766 - 70; PMID: 18600261
- Tsumagari K, Chang S-C, Lacey M, Baribault C, Chittur SV, Sowden J, et al. Gene expression during normal and FSHD myogenesis. BMC Med Genomics 2011; 4:67; http://dx.doi.org/10.1186/1755-8794-4-67; PMID: 21951698
- Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 2010; 5:e15367; http://dx.doi.org/10.1371/journal.pone.0015367; PMID: 21203455
- Jin SG, Jiang Y, Qiu R, Rauch TA, Wang Y, Schackert G, et al. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res 2011; 71:7360 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-11-2023; PMID: 22052461
- Terragni J, Bitinaite J, Zheng Y, Pradhan S. Biochemical characterization of recombinant β-glucosyltransferase and analysis of global 5-hydroxymethylcytosine in unique genomes. Biochemistry 2012; 51:1009 - 19; http://dx.doi.org/10.1021/bi2014739; PMID: 22229759
- Collins CA, Gnocchi VF, White RB, Boldrin L, Perez-Ruiz A, Relaix F, et al. Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PLoS One 2009; 4:e4475; http://dx.doi.org/10.1371/journal.pone.0004475; PMID: 19221588
- Medic S, Rizos H, Ziman M. Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochem Biophys Res Commun 2011; 411:832 - 7; http://dx.doi.org/10.1016/j.bbrc.2011.07.053; PMID: 21802410
- Pane LS, Zhang Z, Ferrentino R, Huynh T, Cutillo L, Baldini A. Tbx1 is a negative modulator of Mef2c. Hum Mol Genet 2012; 21:2485 - 96; http://dx.doi.org/10.1093/hmg/dds063; PMID: 22367967
- Xu YJ, Wang J, Xu R, Zhao PJ, Wang XK, Sun HJ, et al. Detecting 22q11.2 deletion in Chinese children with conotruncal heart defects and single nucleotide polymorphisms in the haploid TBX1 locus. BMC Med Genet 2011; 12:169; http://dx.doi.org/10.1186/1471-2350-12-169; PMID: 22185286
- Bell ML, Buvoli M, Leinwand LA. Uncoupling of expression of an intronic microRNA and its myosin host gene by exon skipping. Mol Cell Biol 2010; 30:1937 - 45; http://dx.doi.org/10.1128/MCB.01370-09; PMID: 20154144
- Shieh JT, Huang Y, Gilmore J, Srivastava D. Elevated miR-499 levels blunt the cardiac stress response. PLoS One 2011; 6:e19481; http://dx.doi.org/10.1371/journal.pone.0019481; PMID: 21573063
- Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ. Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol Rev 2009; 89:1217 - 67; http://dx.doi.org/10.1152/physrev.00017.2009; PMID: 19789381
- Branciamore S, Chen ZX, Riggs AD, Rodin SN. CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc Natl Acad Sci U S A 2010; 107:15485 - 90; http://dx.doi.org/10.1073/pnas.1010506107; PMID: 20716685
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010; 28:495 - 501; http://dx.doi.org/10.1038/nbt.1630; PMID: 20436461
- Dastjerdi A, Robson L, Walker R, Hadley J, Zhang Z, Rodriguez-Niedenführ M, et al. Tbx1 regulation of myogenic differentiation in the limb and cranial mesoderm. Dev Dyn 2007; 236:353 - 63; http://dx.doi.org/10.1002/dvdy.21010; PMID: 17117436
- Havis E, Coumailleau P, Bonnet A, Bismuth K, Bonnin MA, Johnson R, et al. Sim2 prevents entry into the myogenic program by repressing MyoD transcription during limb embryonic myogenesis. Development 2012; 139:1910 - 20; http://dx.doi.org/10.1242/dev.072561; PMID: 22513369
- Ehrlich M, Lacey M. Deciphering transcription dysregulation in FSH muscular dystrophy. J Hum Genet 2012; 57:477 - 84; http://dx.doi.org/10.1038/jhg.2012.74; PMID: 22718021
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011; 10:2662 - 8; http://dx.doi.org/10.4161/cc.10.16.17093; PMID: 21811096
- Kang JS, Krauss RS. Muscle stem cells in developmental and regenerative myogenesis. Curr Opin Clin Nutr Metab Care 2010; 13:243 - 8; http://dx.doi.org/10.1097/MCO.0b013e328336ea98; PMID: 20098319
- Milewski RC, Chi NC, Li J, Brown C, Lu MM, Epstein JA. Identification of minimal enhancer elements sufficient for Pax3 expression in neural crest and implication of Tead2 as a regulator of Pax3. Development 2004; 131:829 - 37; http://dx.doi.org/10.1242/dev.00975; PMID: 14736747
- Brown CB, Engleka KA, Wenning J, Min Lu M, Epstein JA. Identification of a hypaxial somite enhancer element regulating Pax3 expression in migrating myoblasts and characterization of hypaxial muscle Cre transgenic mice. Genesis 2005; 41:202 - 9; http://dx.doi.org/10.1002/gene.20116; PMID: 15789408
- Degenhardt KR, Milewski RC, Padmanabhan A, Miller M, Singh MK, Lang D, et al. Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev Biol 2010; 339:519 - 27; http://dx.doi.org/10.1016/j.ydbio.2009.12.030; PMID: 20045680
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511 - 5; http://dx.doi.org/10.1038/nbt.1621; PMID: 20436464
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5:621 - 8; http://dx.doi.org/10.1038/nmeth.1226; PMID: 18516045
- Tsukamoto K, Nakamura Y, Niikawa N. Isolation of two isoforms of the PAX3 gene transcripts and their tissue-specific alternative expression in human adult tissues. Hum Genet 1994; 93:270 - 4; http://dx.doi.org/10.1007/BF00212021; PMID: 7545913
- Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC, et al. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 2006; 7:67 - 80; http://dx.doi.org/10.1007/s10048-006-0032-6; PMID: 16572319
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 2009; 10:R130; http://dx.doi.org/10.1186/gb-2009-10-11-r130; PMID: 19919682
- Cortese R, Krispin M, Weiss G, Berlin K, Eckhardt F. DNA methylation profiling of pseudogene-parental gene pairs and two gene families. Genomics 2008; 91:492 - 502; http://dx.doi.org/10.1016/j.ygeno.2008.02.004; PMID: 18450418
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 2012; 13:115 - 26; http://dx.doi.org/10.1038/nrm3274; PMID: 22266761
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008; 132:311 - 22; http://dx.doi.org/10.1016/j.cell.2007.12.014; PMID: 18243105
- Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci U S A 2006; 103:7729 - 34; http://dx.doi.org/10.1073/pnas.0600206103; PMID: 16684884
- Zlatanova J, Caiafa P. CCCTC-binding factor: to loop or to bridge. Cell Mol Life Sci 2009; 66:1647 - 60; http://dx.doi.org/10.1007/s00018-009-8647-z; PMID: 19137260
- Myers RM, Stamatoyannopoulos J, Snyder M, Dunham I, Hardison RC, Bernstein BE, et al, ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011; 9:e1001046; http://dx.doi.org/10.1371/journal.pbio.1001046; PMID: 21526222
- Russell MW, Raeker MO, Korytkowski KA, Sonneman KJ. Identification, tissue expression and chromosomal localization of human Obscurin-MLCK, a member of the titin and Dbl families of myosin light chain kinases. Gene 2002; 282:237 - 46; http://dx.doi.org/10.1016/S0378-1119(01)00795-8; PMID: 11814696
- Rubio MD, Johnson R, Miller CA, Huganir RL, Rumbaugh G. Regulation of synapse structure and function by distinct myosin II motors. J Neurosci 2011; 31:1448 - 60; http://dx.doi.org/10.1523/JNEUROSCI.3294-10.2011; PMID: 21273429
- van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell 2009; 17:662 - 73; http://dx.doi.org/10.1016/j.devcel.2009.10.013; PMID: 19922871
- Young P, Ehler E, Gautel M. Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol 2001; 154:123 - 36; http://dx.doi.org/10.1083/jcb.200102110; PMID: 11448995
- Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet 2012; 13:7 - 13; PMID: 22083101
- Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, et al. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol 2012; 8:328 - 30; http://dx.doi.org/10.1038/nchembio.914; PMID: 22327402
- Hackett JA, Zylicz JJ, Surani MA. Parallel mechanisms of epigenetic reprogramming in the germline. Trends Genet 2012; 28:164 - 74; http://dx.doi.org/10.1016/j.tig.2012.01.005; PMID: 22386917
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333:1303 - 7; http://dx.doi.org/10.1126/science.1210944; PMID: 21817016
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011; 146:67 - 79; http://dx.doi.org/10.1016/j.cell.2011.06.020; PMID: 21722948
- Rangam G, Schmitz KM, Cobb AJ, Petersen-Mahrt SK. AID enzymatic activity is inversely proportional to the size of cytosine C5 orbital cloud. PLoS One 2012; 7:e43279; http://dx.doi.org/10.1371/journal.pone.0043279; PMID: 22916236
- Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res 2002; 62:4075 - 80; PMID: 12124344
- Tang H, Araki K, Li Z, Yamamura K. Characterization of Ayu17-449 gene expression and resultant kidney pathology in a knockout mouse model. Transgenic Res 2008; 17:599 - 608; http://dx.doi.org/10.1007/s11248-008-9170-y; PMID: 18288583
- Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 2012; 40:4841 - 9; http://dx.doi.org/10.1093/nar/gks155; PMID: 22362737
- Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, et al. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics 1997; 43:267 - 77; http://dx.doi.org/10.1006/geno.1997.4829; PMID: 9268629
- Bocker MT, Tuorto F, Raddatz G, Musch T, Yang FC, Xu M, et al. Hydroxylation of 5-methylcytosine by TET2 maintains the active state of the mammalian HOXA cluster. Nat Commun 2012; 3:818; http://dx.doi.org/10.1038/ncomms1826; PMID: 22569366
- Sérandour AA, Avner S, Oger F, Bizot M, Percevault F, Lucchetti-Miganeh C, et al. Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res 2012; 40:8255 - 65; http://dx.doi.org/10.1093/nar/gks595; PMID: 22730288
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011; 479:74 - 9; http://dx.doi.org/10.1038/nature10442; PMID: 21964334
- Khare T, Pai S, Koncevicius K, Pal M, Kriukiene E, Liutkeviciute Z, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol 2012; 19:1037 - 43; http://dx.doi.org/10.1038/nsmb.2372; PMID: 22961382
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43 - 9; http://dx.doi.org/10.1038/nature09906; PMID: 21441907
- Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J 2011; 30:4198 - 210; http://dx.doi.org/10.1038/emboj.2011.295; PMID: 21847099
- Zhang Z, Baldini A. Manipulation of endogenous regulatory elements and transgenic analyses of the Tbx1 gene. Mamm Genome 2010; 21:556 - 64; http://dx.doi.org/10.1007/s00335-010-9304-4; PMID: 21107579
- Simrick S, Szumska D, Gardiner JR, Jones K, Sagar K, Morrow B, et al. Biallelic expression of Tbx1 protects the embryo from developmental defects caused by increased receptor tyrosine kinase signaling. Dev Dyn 2012; 241:1310 - 24; http://dx.doi.org/10.1002/dvdy.23812; PMID: 22674535
- Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, et al. Bmp signaling regulates myocardial differentiation from cardiac progenitors through a MicroRNA-mediated mechanism. Dev Cell 2010; 19:903 - 12; http://dx.doi.org/10.1016/j.devcel.2010.10.022; PMID: 21145505
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012; 336:934 - 7; http://dx.doi.org/10.1126/science.1220671; PMID: 22539555
- Brown SJ, Stoilov P, Xing Y. Chromatin and epigenetic regulation of pre-mRNA processing. Hum Mol Genet 2012; 21:R1 R90 - 6; http://dx.doi.org/10.1093/hmg/dds353; PMID: 22936691
- de Wilde J, Hulshof MF, Boekschoten MV, de Groot P, Smit E, Mariman EC. The embryonic genes Dkk3, Hoxd8, Hoxd9 and Tbx1 identify muscle types in a diet-independent and fiber-type unrelated way. BMC Genomics 2010; 11:176; http://dx.doi.org/10.1186/1471-2164-11-176; PMID: 20230627
- Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res 2011; 91:212 - 22; http://dx.doi.org/10.1093/cvr/cvr112; PMID: 21498422
- Zingg JM, Pedraza-Alva G, Jost JP. MyoD1 promoter autoregulation is mediated by two proximal E-boxes. Nucleic Acids Res 1994; 22:2234 - 41; http://dx.doi.org/10.1093/nar/22.12.2234; PMID: 8036150
- Fuso A, Ferraguti G, Grandoni F, Ruggeri R, Scarpa S, Strom R, et al. Early demethylation of non-CpG, CpC-rich, elements in the myogenin 5′-flanking region: a priming effect on the spreading of active demethylation. Cell Cycle 2010; 9:3965 - 76; http://dx.doi.org/10.4161/cc.9.19.13193; PMID: 20935518
- Hupkes M, Jonsson MK, Scheenen WJ, van Rotterdam W, Sotoca AM, van Someren EP, et al. Epigenetics: DNA demethylation promotes skeletal myotube maturation. FASEB J 2011; 25:3861 - 72; http://dx.doi.org/10.1096/fj.11-186122; PMID: 21795504
- Barrès R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab 2012; 15:405 - 11; http://dx.doi.org/10.1016/j.cmet.2012.01.001; PMID: 22405075
- Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet 2011; 7:e1002228; http://dx.doi.org/10.1371/journal.pgen.1002228; PMID: 21852959
- R-Development-Core-Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria: ISBN 3-900051-07-0, 2011.
- Kane MJ, Emerson JW. bigmemory: Manage massive matrices with shared memory and memory-mapped files. 2011; http://CRAN.R-project.org/package=bigmemory.
- Bates D, Maechler M, Bolker B. lme4: Linear mixed-effects models using S4 classes. R package version 0.999375-42. 2011; http://CRAN.R-project.org/package=lme4.
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841 - 2; http://dx.doi.org/10.1093/bioinformatics/btq033; PMID: 20110278
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 2011; 21:1757 - 67; http://dx.doi.org/10.1101/gr.121541.111; PMID: 21750106
- Shibata Y, Sheffield NC, Fedrigo O, Babbitt CC, Wortham M, Tewari AK, et al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet 2012; 8:e1002789; http://dx.doi.org/10.1371/journal.pgen.1002789; PMID: 22761590
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562 - 78; http://dx.doi.org/10.1038/nprot.2012.016; PMID: 22383036