Abstract
Nucleosomes facilitate compaction of DNA within the confines of the eukaryotic nucleus. This packaging of DNA and histone proteins must accommodate cellular processes, such as transcription and DNA replication. The repositioning of nucleosomes to facilitate cellular processes is likely regulated by several factors. In Zea mays, Mediator of paramutation1 (MOP1) has been demonstrated to be an epigenetic regulator of gene expression. Based on sequence orthology and mutant phenotypes, MOP1 is likely to function in an RNA-dependent pathway to mediate changes to chromatin. High-resolution microarrays were used to assay the distribution of nucleosomes across the transcription start sites (TSSs) of ~400 maize genes in wild type and mutant mop1–1 tissues. Analysis of nucleosome distribution in leaf, immature tassel and ear shoot tissues resulted in the identification of three genes showing consistent differences in nucleosome positioning and occupancy between wild type and mutant mop1–1. These specific changes in nucleosome distribution were located upstream as well as downstream of the TSS. No direct relationship between the specific changes in nucleosome distribution and transcription were observed through quantitative expression analysis in these tissues. In silico prediction suggests that nucleosome positioning is not dictated by intrinsic DNA sequence signals in the TSSs of two of the identified genes, suggesting a role for chromatin remodeling proteins in MOP1-mediated pathways. These results also indicate that MOP1 contributions to nucleosome position may be either separate from changes in gene expression, or cooperative with development and other levels of regulation in coordinating gene expression.
Introduction
Packaging of DNA into chromatin allows eukaryotes to contain large volumes of information encoded in DNA sequence into a defined, membrane-bound area of the cell. Chromatin is made up of basic repeating units termed nucleosomes, consisting of two copies each of four types of histone proteins H2A, H2B, H3 and H4. DNA is wrapped approximately 1.7 times around the histone octamer.Citation1 Each nucleosome is separated from the next by linker DNA, which is approximately 38 bp in human,Citation2 18 bp in Saccharomyces cerevisiaeCitation3and 30 bp in Arabidopsis thaliana.Citation4 In maize, linker DNA length genome wide is estimated to be 35 bp.Citation5 The association of DNA with nucleosomes can impact transcription of genes because DNA sequence coding for promoters, enhancers or other regulatory elements could be wrapped around the nucleosome and therefore less accessible to transcription factors.Citation6 Furthermore, following chromatin decondensation and transcriptional initiation, RNA polymerase II progresses through nucleosomally occupied regions during transcriptional elongation.Citation7 However, the relationship between nucleosome position and gene expression is complex, and there is not always a correlation between changes in nucleosome occupancy and transcriptional activity of a gene.Citation8
The positioning of nucleosomes across the genome is determined by multiple factors.Citation9 One of the earliest models is statistical positioning, which involves DNA sequence features acting as boundary constraints to the distribution of nucleosomes.Citation10-Citation12 Statistical positioning is predicted to result in non-random, regular spacing of nucleosomes across a region of DNA; this pattern of nucleosome positioning is referred to as nucleosomal phasing.Citation13,Citation14 Intrinsic DNA sequence properties also play a role in positioning nucleosomes across the genome as sequences have different affinities to the nucleosome core particle.Citation15-Citation17 Chromatin remodelers are another important contributor to nucleosome positioning and several classes of ATP dependent chromatin remodelers have been identified in eukaryotes.Citation18-Citation20 They not only utilize energy from ATP to slide, evict and reposition nucleosomes across the genome,Citation12,Citation21-Citation24 but are also important in the high density assembly of nucleosomes at specific sites across genomes.Citation12,Citation25,Citation26 These factors may act in mutually exclusive or overlapping manners. One potential explanation of the interactions between these is that chromatin remodelers can actively induce changes away from sequence-determined positioning events.
Studies on nucleosome positioning genome-wide, as well as specifically in TSS regions, have been performed in several model organisms including Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans and humans.Citation2,Citation26-Citation29 In A. thaliana, the relationship between DNA methylation and nucleosome positioning has been investigated at a very high resolution,Citation4 while another study in A. thaliana has reported loss of nucleosomes during activation of repetitive elements.Citation30
Herein, we specifically investigate nucleosome distribution across the TSS region of ~400 genes in Z. mays wild type and mediator of paramutation1–1 (mop1–1) mutant tissues, using a high resolution microarray. The mop1–1 mutant was selected because MOP1 has homology to an RNA-dependent RNA polymerase (RDR) thought to participate in RNA-dependent DNA methylation (RdDM).Citation31,Citation32 MOP1 is also known to impact gene expression of many loci,Citation33 and is thought to mediate chromatin structural changes at some regulated loci.Citation34-Citation37 Quantitative expression analysis is also performed to determine the relationship between specific changes in nucleosome distribution and transcription.
Results
Nucleosome position is altered in the transcription start site region of some genes in mop1–1 mutants
Titration of micrococcal nuclease digestion
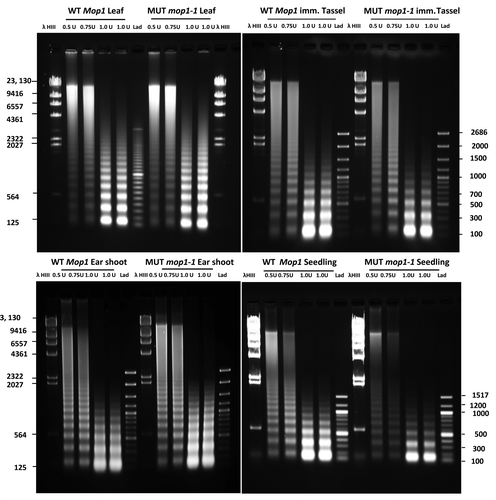
Because DNA associated with nucleosomes is protected from digestion, micrococcal nuclease (MNase) can be used to assay nucleosome position along isolated DNA strands when the chromatin structure is preserved through the extraction process. Chromatin isolated from wild type ear shoot nuclei was used to identify optimal conditions for further experimentation. Increasing the concentration of MNase from 0.0 to 20 U resulted in a gradual change in the pattern of digested fragments of DNA isolated from plant nuclei (Fig. S1). Digested fragments of the size expected for one, two, or multiple nucleosomes and associated DNA, were visible from 0.05 to 0.125 U and a large amount of undigested DNA was detectable as a high molecular weight fraction near the wells. At least eight bands corresponding to oligonucleosomal DNA were visible, while bands ~150 bp in length were observed at concentrations of 0.25 U and higher. This is the approximate size expected for DNA associated with a single nucleosome, and is referred to herein as mononucleosome-sized DNA. At 1.0 U, almost all of the nuclei were digested to some extent (Fig. S1). With 20 U of MNase, most of the nuclei were digested to mononucleosome-sized DNA fragments. Across the range of MNase concentrations, the size of the mononucleosomal DNA decreased gradually, probably due to trimming of the ends of the DNA wrapped around the nucleosomes. The mononucleosomal-sized DNA fragments produced at a concentration of 20 U appear to experience degradation as evidenced by the tailing and smear around the mononucleosome-sized band. Based upon this titration, concentrations of 0.5, 0.75 and 1.0 U were selected for experimental analyses.
Identification of genes with altered nucleosome distribution in transcriptional start site adjacent regions
To identify tissue-specific examples of chromatin changes in mop1–1 mutants, nucleosome positions were determined in seedling, leaf, ear shoot, and tassel of wild type and homozygous mop1–1 plants. Distinct nucleosomal ladders were obtained from all four tissue types using three concentrations of MNase (). Nuclei derived from leaf tissue appeared more resistant to nuclease digestion based on the intensity and size of the bands corresponding to mono-/di-nucleosomal DNA, and the presence of high molecular weight DNA in the gel. For the remaining three tissue types, a high proportion of DNA extracted from nuclei was digested to mono-/di-nucleosomal fragments. At MNase units 0.5 and 0.75, a smear preceding the ladders was present higher up in the gel for all four tissue types. Most, if not all, of the smear disappeared at 1.0 U, except for leaf tissue where mono-/di-nucleosomal fragments were less abundant.
Figure 1. Agarose gel fractionation of MNase digested DNA from leaf, immature tassel, ear shoot and seedlings of homozygous wild type (WT MOP1) and homozygous mutant mop1–1 (MUT mop1–1) plants. For each genotype and tissue type, separate reactions were conducted using 0.5, 0.75, and 1 unit of MNase respectively. Lanes with molecular weight markers are indicated (λ HindIII and Lad); the size of each band is indicated in base pairs.
The mop1–1 mutation was originally isolated in laboratory specific genetic stocks, consisting primarily of K55 and W23 inbred lines and active Mutator stocks.Citation34 This particular genetic background has not been sequenced in its entirety, but Zea mays as a species is known for its genetic diversity.Citation38 Structural diversity in distinct maize haplotypes results in differential hybridization behaviors that are detectable using microarray analysis techniques, and these comparative genome hybridizations have been informative in characterizing different inbred lines of maize.Citation39
The plant material utilized for nucleosome distribution analysis resulted from multiple generations of backcrossing to introgress the mop1–1 mutation into the B73 reference genome, but as mop1–1 and other associated genetic loci related to plant pigmentation were continuously selected for, a certain amount of genetic variability persisted in segregating populations. We used comparative genome hybridization (CGH), with the same custom microarray used to analyze nucleosome position, to identify genomic regions in the mop1–1 introgression lines that had variable hybridization behavior between wild type and mutant plants. It is anticipated that these genomic regions would contain genetic variability between the original mop1–1 stock and the B73 reference genome, and represent linkage drag associated with recurrent selection for the mop1–1 allele and its associated pigmentation phenotype in segregating populations. Such regions would be unsuitable for nucleosome position analysis by microarray, because differential hybridization due to nucleosome association could not be distinguished from differential hybridization due to genetic differences.
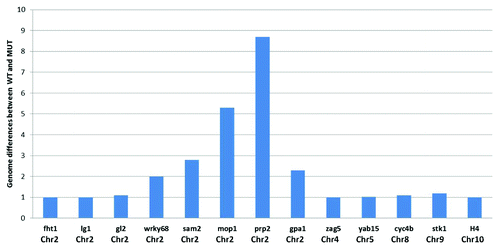
The CGH plots identified genetically distinct regions between mutant and wild type individuals in the same segregating family. The magnitude of the differences between the genomic regions (wild type vs. mutant mop1–1) was calculated from the log2 ratio of the signal intensity. A pronounced difference between the genome of wild type and mutant mop1–1 was observed around the chromosomal location of the mop1–1 gene (). This region, and others exhibiting evidence of genetic variability between the two genotypes, were excluded from further analysis. Of the 399 genes on the array, nucleosome position in the TSS region could be analyzed for 382 of them.
Figure 2. Schematic representation of comparative genomic hybridization (CGH) on the 400 gene TSS microarray for 13 exemplar genes with their respective chromosomal locations. The magnitude of sequence differences (ratio of the log2 ratios of signal intensity for respective samples) between wild type and mutant mop1–1 is displayed on the y axis for each gene. A value of 1 in the bar chart indicates that there is no detectable difference in hybridization behavior between the genomic DNA from wild type and mutant mop1–1 individuals in the probed region for the gene.
Genomic regions not excluded by CGH were analyzed for differences in nucleosome distribution between replicates of wild type and mutant mop1–1. By observation of R-generated plots, the nucleosome distribution around the TSS for each gene was compared between each replicate of mutant and wild type leaf, ear shoot, tassel and seedling samples. These tissues were selected because mop1–1 mutants exhibit developmental abnormalities, suggesting that MOP1 activity might be important during multiple developmental stages.Citation34 For three genes, flavanone 3−hydroxylase1 (fht1), liguleless1 (lg1), and yabby15 (yab15), consistent differences in nucleosome organization between mutant and wild type replicates were observed in one or more of the tissues analyzed (). Two of the genes, fht1 and lg1, are located on chromosome 2, while yab15 is located on chr5. No differences were observed in seedling tissues, and this tissue was not used in subsequent analyses. For fht1 and lg1, nucleosome position varied both up and downstream of the annotated transcription start site of the gene, and the most consistent differences reflected a loss of a positioned nucleosome in mutant mop1–1 plants. Differences in nucleosome position in the TSS region of fht1 were observed in ear shoot tissue, while consistent differences in nucleosome position in the TSS region of lg1 were observed for leaf, immature tassel, and ear shoot tissues. The nucleosome distribution profile of the lg1 TSS was similar, although not identical for all three tissues (Fig. S2), and the observed differences between mutant and wild type were consistent in all three tissues for this gene. A high density of repetitive DNA elements prevented microarray analysis of the region upstream of the yab15 TSS (data not shown). The region downstream of the annotated TSS was amenable to analysis, and consistent differences were observed in the region downstream of the annotated TSS of yab15 in immature tassel. For this gene, the consistent difference reflects an increased abundance of positioned nucleosomes in a limited region of the analyzed sequence in mutant individuals.
Figure 3. Nucleosome distribution plots for 3 genes exhibiting differences between wild type and mop1–1 homozygous mutants. The x-axis shows the coordinates of the genes flavanone 3-hydroxylase1 ((A), liguleless1 (B), and yabby15 (C) on their respective chromosomes, while the y-axis represent the log 2 ratio of the signal intensity between the test (nucleosomal DNA) and reference (genomic DNA) samples. The black lines represent replicates of wild type MOP1, while the red lines represent replicates of mutant mop1–1. The location and direction of transcription is indicated with an arrow and label for each gene. Open arrows indicate regions where nucleosome occupancy appears to vary between wild type and mutant plants. For yabby15 and liguless1, the plots represent a comparison of wild type and mutant immature tassel samples. For flavanone 3-hydroxylase1, the plot represents a comparison of wild type and mutant ear shoot samples.
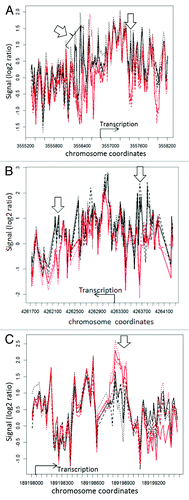
The average covariance between mutant and wild type for each of these three genes were compared (), and the averaged r-value for mutant compared with wild type was notably lower than that for wild type compared with wild type, and mutant compared with mutant, for the ear shoot samples at the TSS of fht1, and the leaf, tassel and ear shoot samples of lg1. The average r-values for wild type compared with wild type, mutant compared with mutant, and wild type compared with mutant were not substantially different at the TSS of yab15 in any tissues, although there were consistent differences between genotypes in the immature tassel samples observable in the nucleosome distribution plots (). The average r-values in the TSS of yab15 for mutant compared with mutant, mutant compared with wild type, and wild type compared with wild type were lower than those calculated for the other two genes, suggesting a higher level of variability in nucleosome distribution in some portions of this locus, which is also observable in the nucleosome distribution plot ().
Table 1. Genes showing consistent differences in nucleosome distribution between wild type and mop1–1 mutants
Nucleosome positioning is not predicted by sequence signals in the TSSs of fht1, lg1, and yab15
One determinant of nucleosome position can be signals intrinsic to DNA sequence. Support vector machines (SVM) have been utilized to predict nucleosome-forming and nucleosome-inhibitory sequences in genomic sequences.Citation40 An SVM generated using empirical nucleosome distribution data from human chromatin has been generated and applied to the maize genome sequence (www.maizenucleosome.org/data/nol/), resulting in genome wide predictions of nucleosome occupancy likelihoods (NOL) based upon DNA sequence features (Fincher, Dennis and Bass, personal communication). Across the entire data set of 382 genes, the NOL SVM predicts the observed nucleosome positioning in our data sets with varying degrees of accuracy, and exhibits an average r-value of 0.458 across all tissues when compared with wild type samples, and an average r-value of 0.446 when compared with mutant samples. To determine whether nucleosome occupancy at the TSSs of fht1, lg1, and yab15 coincided with SVM-predicted nucleosome-forming and -inhibitory sequences, the distribution of nucleosomes in these regions were compared with the NOL SVM predictions in a pairwise fashion using R (). For each gene, the average r-values were similar if averages were compared by tissue type and genotype, suggesting that the relationship between empirically determined nucleosome distribution and sequence-based predictions does not differ between wild type and mutant or the analyzed tissues. Of the three genes, fht1 had the highest r-values for comparisons of nucleosome position data with the NOL SVM predictions, with r-values between 0.58 and 0.71 across the different tissue types. These values were higher than those observed across all data points for each sample, for which r-values ranged from 0.37 to 0.508.
Table 2. Comparison between observed nucleosome distribution and support vector machine predicted nucleosome-forming and -inhibitory sequences in the TSSs of three genes with altered nucleosome distribution in mop1–1 mutants
Changes in gene expression are not coincident with changes in nucleosome distribution in mop1–1 mutants
Because nucleosome distribution within the TSS region can be related to the expression level of the associated gene, quantitative expression analysis was used to assay expression of liguleless1, flavanone 3−hydroxylase1, yabby15 and colored plant1 (b1). The b1 (colored plant1) gene was included because mop1–1 plays a role in its transcriptional silencing, and this gene is upregulated in mop1–1 mutants.Citation31,Citation34,Citation41 The four genes were not expressed at uniform levels across all tissue types (Fig. S3), and so expression was compared between mutant and wild type samples in each tissue individually. As expected, the b1 gene was upregulated in mutant mop1–1 ear shoot and immature tassel ( = 0.16 for ear shoot and p = 0.14 for tassel using two tailed paired t-test). For leaf tissue, b1 transcripts were not detected in wild type individuals and were only detected in mutant mop1–1 samples at high CT values, consistent with the phenotypic expression pattern of B1-induced pigmentation in the plants used for this analysis (data not shown).
Figure 4. Real time PCR results for flavanone 3-hydroxylase1 (A) liguleless1 (B) yabby15 (C), and colored plant1 (D). Transcript abundance was measured in leaf, tassel and ear shoot of mop1–1/mop1–1 mutant (MUT) and wild type MOP1/MOP1 (WT) plants using ubiquitin as an endogenous control. Relative quantification (Rq) values were averaged for three replicates of mop1–1 samples for each tissue type, and compared with the average value for three replicates of wild type samples (set at 1 for each tissue type). The averaged Rq values are plotted on the y-axis. Error bars represent standard error for the averaged Rq values.
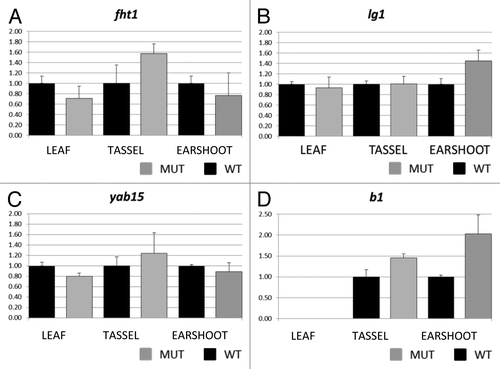
In mop1–1 mutants, fht1 was upregulated in tassel, and consistent genotype specific differences in expression were not observed in ear shoot and leaf tissues. The gene liguleless1 appeared upregulated in ear shoots ( = 0.143 using two tailed paired t-test), and genotype specific differences were not observed for leaf and immature tassel. For yabby15, expression was slightly lower in wild type leaves compared with mutant leaves, but no consistent genotype specific differences were observed in other tissues.
Discussion
This analysis identified 3 genes from a total of 382 in the data set with consistent changes in nucleosome occupancy and positioning in mutant compared with wild type plants. This demonstrates that in plants that lack wild type MOP1 protein, some changes in nucleosome positioning occur around gene transcription start sites. The mechanisms that induce the changes in nucleosome position are not known at this time. One possibility is that in wild type plants, MOP1 directly or indirectly mediates nucleosome position at the TSS of some genes, and this positioning becomes disrupted in the absence of functional MOP1. Another possibility is that the lack of a functional MOP1 protein induces changes in the level of transcriptional activity, and nucleosomes are repositioned directly or indirectly as a result of differential RNA polymerase II activity.
Prior to this analysis, a change in nucleosome distribution related to changes in expression had been demonstrated for one known target of MOP1 mediated regulation, b1.Citation35 Epigenetic regulation of B’, a paramutagenic allele of b1, by MOP1 has also been previously demonstrated.Citation34 The regulatory regions for this gene are located 100kb upstream of the promoter proximal and are not tightly linked to the TSS.Citation37 This regulatory locus is not amenable to the analysis techniques utilized for this study, due to its repetitive nature and the fact that its chromosomal location is within the region excluded from this analysis by comparative genome hybridization results, so direct comparisons cannot be made. However, if the 382 genes included in this study are representative of all genes in the maize genome, ~0.7% of genes might be expected to demonstrate nucleosome distribution changes in mop1–1 plants. Thus, expanding this analysis might facilitate the identification of additional genomic loci that are regulated by MOP1-mediated mechanisms.
A genome wide study of nucleosome positioning in S. cerevisiae demonstrated that ~50% of nucleosome organization across the genomes can be explained by intrinsic DNA sequence.Citation17 Recent studies in S. cerevisiae and S. pombe have shown the importance of chromatin remodelers in organization and maintenance of nucleosome structure. Mutations in several ATP-dependent chromatin remodeling enzymes in S. cerevisiae and S.pombe, including the chromodomain-helicase-DNA-binding protein1 (CHD1), result in a loss of nucleosome organization across the TSS and elsewhere in the genome.Citation12,Citation18-Citation20 It is therefore possible that the gene-specific profile of nucleosome organization observed across TSS is the result of DNA sequence and chromatin remodeling enzymes. An SVM trained with empirical data was used to predict nucleosome distribution in the TSSs of the three genes with changes in nucleosome distribution in one or more tissue of mop1–1 mutants. This analysis revealed a very low correlation between predicted and observed nucleosome occupancy in these TSS flanking regions for yab15 and lg1. This suggests that DNA sequence signals may not be the predominant determinate of nucleosome position in these regions, and might indicate a role for chromatin remodelers in establishing and maintaining nucleosome position in these regions. Based on its orthology with Arabidopsis RDR2 and phenotypes for maize mutants, the current model for MOP1 activity is that it functions in a small RNA mediated pathway to direct DNA methylation and heterochromatin formation at transcriptionally silenced loci.Citation31-Citation33,Citation42,Citation43 An association of chromatin remodeling proteins with MOP1 activity is therefore consistent with the predicted function and mechanism of MOP1 activity, but further testing is required to determine whether this type of activity is involved in the nucleosome position changes observed in this study.
Another predicted hallmark of MOP1 activity is transcriptional silencing, and the expression pattern of many genes changes in some tissues of mop1–1 mutants.Citation33 Results from our study indicated that fht1 was slightly upregulated in immature tassels of mop1-1, but not in ear shoots, where nucleosome distribution changes were observed. Lg1 was upregulated in only one of the tissues where nucleosome distribution changes were observed. Expression of yab15 was slightly lower in the leaves of mop1–1 mutants, where there was not an apparent change in nucleosome distribution, and no change in expression was detected in immature tassel, where nucleosome distribution changes were observed in mutant relative to wild type.
These results do not support a direct causal relationship between the specific changes in nucleosome distribution observed across TSS and transcription of the associated gene. It is possible that changes in nucleosome position do not dictate, but rather predispose, a locus to changes in expression at a different developmental stage or in response to another signal. For example, fht1 has been genetically demonstrated to be regulated by a transcription factor, R1, with tissue specific activity.Citation44 Both R1 and B1 are basic helix loop helix transcription factors that have been demonstrated to be functionally redundant in maize.Citation45 MOP1 is known to epigenetically upregulate some alleles of b1 and r1,Citation34 and our qRT-PCR analysis confirmed upregulation of b1 in tassel and ear shoot tissues of homozygous mutant individuals. Thus, the observed upregulation of fht1 might be caused by upregulation of b1of b1 in the tassels of mop1–1 mutants. Upregulation of b1 was also observed in ear shoot samples of mop1–1 homozygous individuals, but fht1 did not appear transcriptionally upregulated in mutant ear shoots. The regulation of genes in the flavonoid/anthocyanin biosynthetic pathway requires multiple transcription factors, and it is also possible that another required factor for fht1 upregulation is not present in the ear shoots.
Of the three genes described in this study, two genes exhibit very low correlation between nucleosome distribution and predicted sequence signals, which may be indicative of the involvement of chromatin remodeling proteins in nucleosome positioning at these loci. Because MOP1 is known to be an epigenetic regulator of genes that demonstrate changes in nucleosome distribution during regulated changes in gene expression,Citation35 nucleosome distribution may be misregulated in mop1–1 mutants due to the disruption of a MOP1-mediated chromatin-remodeling event at the TSSs of Yab15 and Lg1. Nucleosome distribution in the TSS region of Fht1 exhibits a higher degree of correlation with predicted sequence signals for nucleosome distribution. It is possible that this result reflects an indirect relationship between loss of a functional MOP1 protein and changes in chromatin structure at the fht1 locus, mediated by the transcription factor B1, and other potential interacting proteins. Expression and siRNA sequence analysis of mop1–1 homozygous individuals has indicated other evidence for primary and secondary effects of MOP1 in maize, including a wide range of differentially expressed genes with examples of upregulation and downregulation, the presence of MOP1-dependent and -independent small RNAs, and changes in the expression level of many predicted chromatin-related proteins that might be expected to change expression of other genes.Citation33 This would imply that some mop1–1 related phenotypes result from a loss of MOP1 activity at a specific locus, and that these direct changes may trigger downstream effects that are detectable as secondary effects in mop1–1 homozygous mutants.
Another possibility is that the changes in transcriptional activity and changes in nucleosome distribution in mop1–1 mutants are induced by discrete mechanisms, and not related to one another. The relative number of genes exhibiting changes in nucleosome distribution in mop1–1 mutants is notably lower than the number of genes that are thought to be transcriptionally misregulated in mop1–1 mutants, reported to be as high as 6000 genes in shoot apical meristems.Citation33 Thus, the results of this study imply that changes in nucleosome distribution in the TSS region are not necessarily a unifying feature of all loci that exhibit MOP1-associated transcriptional changes, but that loss of a functional MOP1 protein does result in observable and discrete changes in nucleosome distribution at some loci.
This approach identified three genes with differential nucleosome distribution in maize mop1–1 mutants, suggesting a role for RNA-directed mechanisms in nucleosome positioning at some loci. These changes in nucleosome distribution were not directly associated with changes in gene expression. For two of the genes, the nucleosome distribution changes do not appear to be related to intrinsic DNA sequence signals in the affected regions, suggesting the involvement of trans-acting factors, like chromatin remodeling proteins, in positioning nucleosomes in these regions. These results imply that changes in chromatin structure may not always be related differential gene expression and may have other functions or implications. This could be an indication of changes in gene expression under multiple layers of regulation, making changes in nucleosome position temporally or developmentally separate from the transcriptional changes.
Materials and Methods
Plant materials
Maize B73 seeds were obtained from the Maize Genetics Cooperation Stock Center (http://maizecoop.cropsci.uiuc.edu/). The mutant mop1–1 alleleCitation34 in W23 and K55 background was introgressed into B73. Plants used in the current study were derived from a sixth-generation backcross with B73 as the recurrent parent (BC6), and grown in a greenhouse (Department of Biological Science, Florida State University) under controlled conditions. Immature tassels ~5–10 cm in length were excised from the plants using razor blades. The immature tassels were measured, immediately placed in individual 15 ml conical tubes and flash frozen in liquid nitrogen. Ear shoots ~5 cm in length were also harvested by excising developing ears from plants with a razor blade. Husk leaves were removed and the tissue was flash frozen as above. At the time of ear and tassel collection, one leaf from each plant was also collected for genotyping. Ten day-old seedlings were also harvested from by removing the seedlings from the soil and cutting away the below-ground tissue with a razor blade. The rootless seedlings were placed in individual envelopes, and flash frozen in liquid nitrogen. All BC6 progeny assayed were derived from the same parental mutant mop1–1 individual.
DNA extraction and genotyping
Genomic DNA for the first BC6 segregating family was extracted using the Mini-CTAB protocol.Citation46 For seedlings, genomic DNA was isolated using a fast modified version of Mini-CTAB protocol using the Mixer Mill (Retsch®). Briefly, one leaf disc was placed in each of the 96 racked collection microtubes (Qiagen) and 100 µl of CTAB isolation buffer [2% w/v CTAB, 1.4 M NaCl, 0.2% v/v β-mercaptoethanol, 20 mM EDTA and 100 mM TRIS-HCl (pH 8.0)] was added to each tube. One stainless steel bead (Qiagen) was placed in each tube prior to grinding for 2 min at 30 Hz using the Mixer Mill. Following grinding, 500 µl of CTAB was added to each sample and the homogenate was ground again for 15 sec at 30 Hz. The samples were then incubated for 30 min at 65°C in an incubator. After a brief centrifugation at 300 xg for 3 min, 300 µl of chloroform was added to each tube. The homogenate was mixed with chloroform (EMD Biosciences) by inverting the tubes several times before centrifugation at 3000 xg for 5 min at RT. The supernatant was transferred into 1.5 ml tubes and 500 µl of cold isopropanol was added to each tube. Precipitation of DNA was performed at 14,000 xg for 5 min at 4°C. The pellet was allowed to air-dry overnight and resuspended in 50 µl of ddH20.
Genomic DNA extracted from each plant was used for mop1–1genotyping using primers KM384F (TCTCCACCGCCCACTTGAT), KM385R (CCCAAGAGCTGTCTCGTATCCGT) and KM386R (CTTCATCTCGAAGTAGCGCTTGTTGTCC). Each plant was genotyped with three different primer combinations to accurately determine the genotypes for mop1–1 wild type and mutant alleles. PCR conditions were as follows: initial denaturation 5 min at 94°C followed by 30 cycles of denaturation at 95°C 35 sec, annealing at 56°C 45 sec and extension at 72°C for 45 sec. A final extension for 10 min at 72°C was included. The amplification products were run on a 1.5% agarose gel for 1 h 15 min. Amplicons were visualized using the Gel doc imaging system (Bio-Rad), and progeny were scored for their genotypes for mop1–1. Plants homozygous for MOP1 wild type as well as those homozygous for the mutant mop1–1 allele were selected for nuclei isolation.
For use as a reference and comparative genomic DNA hybridization (CGH), genomic DNA was extracted using the DNeasy Plant Maxi kit (Qiagen) following the manufacturer’s protocol. Progeny used for genomic DNA extraction were derived from the same segregating BC6 family.
Nuclei isolation
Leaves, immature tassels, ear shoots, and seedlings obtained from homozygous wild type and mutant mop1–1 progeny, were ground separately to a fine powder in liquid nitrogen using mortar and pestle. Samples from wild type and mutant plants were pooled separately to obtain enough material for nuclei isolation for each replicate. A total of three replicates were analyzed for each tissue and genotype with the exception of seedlings, where only two replicates were included due to very low yield of nuclei from this tissue type. Leaf samples from four individual plants were pooled per replicate for each genotype; immature tassels from three individual plants were pooled per replicate for each genotype; ear shoots from two wild type individual plants were pooled for wild type and ear shoots from individual plants rather than pools were used for each replicate for mutants; six individual seedlings were pooled per replicate for each genotype. Nuclei isolation was performed using APEL buffer (20mM TRIS-HCl pH 7.8, 250mM sucrose, 5mM MgCl2, 5mM KCl, 40% glycerol, 0.25% Triton X-100 0.1% BME).Citation47 Ground tissue was kept cold in liquid nitrogen until fixation. Approximately 10 g of ground material was used for every 10 ml of APEL buffer. To 18 ml of nuclei isolation buffer, 2 ml of formaldehyde was added and the ground material was mixed vigorously with APEL buffer using a magnetic stirrer until a homogenate was obtained. The suspension was incubated for 10 min at RT with occasional stirring. Following fixation, 1 ml of 2.5 M glycine was added to the suspension with slow stirring. After incubation for 5 min at RT, the suspension was poured into 50 ml conical tubes and centrifuged at 2000 xg for 10 min at 4°C. The supernatant was carefully poured off and the pellet resuspended in 20 ml APEL buffer containing 50 µl of Triton X-100 (EMD Biosciences) and 18 µl of β-mercaptoethanol. The suspension was allowed to incubate on ice for 5 min, and filtered through 4 layers of pre-wetted cheesecloth placed on a funnel. The filtrate was centrifuged at 2000 xg for 10 min at 4°C. The pellet was resuspended as above and the centrifugation step was repeated to further eliminate plant debris. Final resuspension of the isolated nuclei was performed in 1 ml of microccocal nuclease (MNase) digestion buffer (50 mM TRIS-HCl pH 7.5, 320 mM sucrose, 4 mM MgCl2, 1 mM CaCl2). The suspension was mixed with 1 µl of 0.1M PMSF (EMD Biosciences) and the samples were stored at -80°C.
Quantification of nuclei
The amount of DNA in the isolated nuclei samples was quantified using lambda DNA as a reference. A portion of the isolated nuclei sample was subjected to decrosslinking (see below) for 6+ hrs followed by a phenol-chloroform extraction. Briefly, a volume of 50 µl of isolated nuclei was aliquoted for each sample, except for nuclei derived from seedling where a volume of 100 µl was used. Two samples containing 10 µg of lambda DNA were also included in the set. The volume was adjusted to 100 µl using microccocal nuclease digestion buffer, and 100 µl of ddH20 was added to each tube. Following addition of Proteinase K (Amresco) and 10% SDS (Mallinckrodt Baker Inc.), the samples were decrosslinked overnight at 65°C. The samples were then subjected to a phenol-chloroform extraction. Final DNA resuspension was performed in ~30 µl of ddH20. The amount of DNA in each sample was quantified using a Nanodrop spectrophotometer (Thermoscientific). The percent of lambda DNA recovered during phenol-chloroform extraction was estimated using the [(x/10)*100] where x is the yield of lambda DNA in microgram. The original amount of DNA in each sample of the 1 ml of isolated nuclei was then estimated assuming a 100% recovery rate from the two lambda DNA samples using the formula [(w/y)*100] where w is the yield of DNA from the 30 µl of resuspended DNA, and y is the percent lambda DNA recovered. Using the value obtained, an estimate of the amount of DNA in the 1 ml of nuclei was calculated by multiplying a by 20 for the volume of nuclei sample initially used for quantification.
Micrococcal nuclease (MNase) Digest
To determine the units of MNase to use for this study, ~8 µg nuclei isolated from a B73 ear shoot was subjected to a 4 min nuclease digestion with an increasing concentration of MNase. According to the pattern obtained, concentrations of 0.5 units (U), 0.75 U and 1.0 U of MNase were selected for experimentation. For MNase digest of mop1–1 nuclei for each replicate, the isolated nuclei were aliquoted into four tubes each containing ~6 µg of DNA. The volume of nuclei samples was then adjusted to 100 µl using microccocal nuclease digestion buffer. To each of the four nuclei samples (~6 µg), 2 µl of 20 mg/ml Proteinase K was added followed by 20 µl of 10% SDS. After mixing vigorously, the samples were incubated at 65°C for 6+ hrs in a heat block for decrosslinking. DNA was then isolated using phenol-chloroform extraction as above.
Purification of mono-/di-nucleosomal DNA by freeze-squeeze method
The DNA fragments resulting from each nuclei digest were fractionated by electrophoresis in a 1.0% agarose gel for 2 h. Post-staining was performed for 30–45 min in 1 L of ddH20 containing 50 µl of ethidium bromide (10 mg/ml). The gel was then de-stained in 1L of ddH20 for 45 min. Nucleosomal ladders were visualized using the Gel Doc imaging system.
Mono- and di-nucleosomal DNA was excised from the gel using razor blades and purification of the nucleosomal DNA fragments was performed using a modified freeze-squeeze methodCitation48 and 5 mL syringes without needles (BD Biosciences). Each gel slice was inserted into an open syringe containing multiple layers of Miracloth (Calbiochem) inside a 15 ml conical tube (VWR), and stored overnight in a -80°C freezer. Following freezing, the gel slices were allowed to thaw for 15–20 min at RT before centrifuging at 3000 xg for 30 min at RT. The liquid recovered was then transferred to 1.5 ml microcentrifuge tubes and 1 µl of linear polyacrylamide (EMD Biosciences) was added to each sample to maximize the recovery of nucleosomal DNA. Two volumes of 100% ethanol were then added to each tube, and the samples were placed in a -20°C freezer for at least 20 min. The precipitated DNA was centrifuged at 18,000 xg for 20 min at 4°C. The pellet was washed with 70% ethanol by centrifuging at 18,000 xg for 5 min at 4°C. The samples were then placed in a Savant SpeedVac® concentrator (Thermo Electron Corporation) to remove residual ethanol. Resuspension of the pellet containing mono- and di-nucleosomal DNA was performed in 40 µl of ddH20.
Description of Transcription Start Site microarray
The tiling transcription start site array was based on sequences of ~400 genes, most of which are classical, or frequently studied, genes from the COGE website (http://genomevolution.org/wiki/index.php/Classical_Maize_Genes). A custom 12-plex TSS array (12x135k) consisting of 12 sub-arrays was manufactured by NimbleGen. Sequences ~1500 bp upstream as well as downstream of the TSS were downloaded from www.maizegdb.org/ (B73 RefGen_v2), and used to design isothermal probes 50–75 bp in length across the TSS for each of the selected genes at 15 bp intervals (Table S1). For some genes, the sequence in the 3000 bp window around the TSS region included sequences with more than 10 copies in the genome. These highly repetitive probes were not included on the array, and probe number was reduced for those genes.
Labeling and probing of microarray
Purified mono- and di-nucleosomal DNA from wild type and mutant mop1–1 from each of the four tissue types along with genomic DNA samples were provided to the Molecular Core Facility (Department of Biological Sciences, Florida State University). Mono-/di-nucleosomal DNA (test) was fluorescently labeled with Cy3, while genomic DNA (reference) was labeled with Cy5 using the NimbleGen dual color DNA labeling kits following the manufacturer’s protocol (NimbleGen). A test and a reference sample were hybridized to each sub-array.
To perform comparative genomic hybridization (CGH), genomic DNA from cy3-labeled mutant mop1–1 sample was compared with a cy5-labeled wild type mop1–1 sample. The hybridization pattern and intensity of signals from bare genomic DNA derived from wild type and mutant mop1–1 samples were also investigated by comparing the ratio of two cy3-labeled mutant samples to two cy5-labeled wild type mop1–1 samples. Four individuals were represented in each of the two replicates used for genomic DNA hybridization. Probing of the array was performed following the manufacturer’s instructions (NimbleGen).
Generating nucleosome distribution (ND) plots with R
Following microarray, data were extracted and processed using the DEVA software (NimbleGen), where alignment, normalization of the ratios and segmentation analysis were performed. The GFF files containing the processed data from the TSS array, were analyzed using the software R (R Development Core Team). The commands from http://chromatin.bio.fsu.edu/ were used for generating the ND plots from the microarray data.
RNA isolation and Real Time PCR
RNA was isolated from leaf, immature tassel and ear shoot using TRI REAGENT® (Molecular Research Center, Inc.) following the manufacturer's instructions. The isolated RNA was treated with RQ1 RNase Free DNase (Promega) for 30 min at 37°C prior to purification using the RNA Clean and Concentrator Kit (Zymo Research). Total RNA was eluted in 25 µl of RNase Free water and quantified using the Nanodrop spectrophotometer. cDNA was synthesized using the SuperScript III First-Strand Synthesis system (Invitrogen) following the manufacturer’s instructions. Primers were designed for three genes: liguleless1 (Lg1), flavanone 3-hydroxylase1 (Fht1) and yabby15 (Yab15) using the software Oligo Explorer.
Primer efficiency tests were performed on different sets of primers prior to the qRT-PCR assay on cDNA synthesized from the WT leaf tissue. The forward and reverse primer used to analyze expression of each gene by qRT-PCR were:
Yab15F2 CAGCAGCCTGTTCAAGACGGTGAC
Yab15R2 GAGTGAGCGAAGTTGAGATGGTTG
Iig1F2 CCAGACCCAAGCCGTCTCCAGTGA
Iig1R2 CGAGCTGCAAGATGTTGCTGTGTT
Fht1F3 ATTCTTGCCTGATATGGTAGGGGG
Fht1R3 TAATCACCACACGGTCGCACGTAG
UbiqF GACTACACGATGGAGAACATCCTAA
UbiqR GAAGAATGTCCCTTCTGGAGGCTG
b1F1 AGGAGCTTCAACGAAGGGTACAAG
b1R1 GGAGTTGGAGCCCACACAGACTTT
The primer efficiencies were 122% for Yab15; 105% for Fht1; and 92% for Lg1. All RT-PCR reactions were performed by the Molecular Core Facility (Department of Biological sciences, Florida State University) using SYBR® Green PCR Mastermix (Applied Biosystems) on the 7500 Fast Real Time PCR Systems (Applied Biosystems). The comparative CT method was used to calculate the fold changes in expression.
Additional material
Download Zip (714.6 KB)Acknowledgments
We would like to thank the Florida State University Department of Biological Science Molecular Core Facility, especially Steve Miller, and Brian Washburn for their assistance with microarray analysis and qRT-PCR, respectively. We are also grateful to Gregg Hoffman for his valuable help during harvesting of seedlings. We would like to thank Thelma Madzima for assistance with propagation and collection of tissue from the appropriate genetic stocks, and for reading and commenting on the manuscript. We also wish to thank Theresa Jepsen for her help with plant growth, Loury Migliorelli for technical assistance and helpful discussion over the course of the project and H.W. Bass and J.H. Dennis for discussion and data analysis assistance. J. Kennedy provided editorial assistance with the manuscript. This work was funded by the National Science Foundation Plant Genome Research Program through award IOS-PGRP-1025954.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999; 98:285 - 94; http://dx.doi.org/10.1016/S0092-8674(00)81958-3; PMID: 10458604
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008; 132:887 - 98; http://dx.doi.org/10.1016/j.cell.2008.02.022; PMID: 18329373
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 2007; 39:1235 - 44; http://dx.doi.org/10.1038/ng2117; PMID: 17873876
- Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010; 466:388 - 92; http://dx.doi.org/10.1038/nature09147; PMID: 20512117
- Gent JI, Schneider KL, Topp CN, Rodriguez C, Presting GG, Dawe RK. Distinct influences of tandem repeats and retrotransposons on CENH3 nucleosome positioning. Epigenetics Chromatin 2011; 4:3; http://dx.doi.org/10.1186/1756-8935-4-3; PMID: 21352520
- Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A 2007; 104:12955 - 61; http://dx.doi.org/10.1073/pnas.0704138104; PMID: 17670940
- Kornberg RD. The molecular basis of eucaryotic transcription. Cell Death Differ 2007; 14:1989 - 97; http://dx.doi.org/10.1038/sj.cdd.4402251; PMID: 18007670
- Huebert DJ, Kuan PF, Keleş S, Gasch AP. Dynamic changes in nucleosome occupancy are not predictive of gene expression dynamics but are linked to transcription and chromatin regulators. Mol Cell Biol 2012; 32:1645 - 53; http://dx.doi.org/10.1128/MCB.06170-11; PMID: 22354995
- Radman-Livaja M, Rando OJ. Nucleosome positioning: how is it established, and why does it matter?. Dev Biol 2010; 339:258 - 66; http://dx.doi.org/10.1016/j.ydbio.2009.06.012; PMID: 19527704
- Kornberg RD, Klug A. The nucleosome. Sci Am 1981; 244:52 - 64; http://dx.doi.org/10.1038/scientificamerican0281-52; PMID: 7209486
- Fedor MJ, Lue NF, Kornberg RD. Statistical positioning of nucleosomes by specific protein-binding to an upstream activating sequence in yeast. J Mol Biol 1988; 204:109 - 27; http://dx.doi.org/10.1016/0022-2836(88)90603-1; PMID: 3063825
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 2011; 332:977 - 80; http://dx.doi.org/10.1126/science.1200508; PMID: 21596991
- Kornberg RD, Stryer L. Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism. Nucleic Acids Res 1988; 16:14A 6677 - 90; http://dx.doi.org/10.1093/nar/16.14.6677; PMID: 3399412
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 2008; 18:1073 - 83; http://dx.doi.org/10.1101/gr.078261.108; PMID: 18550805
- Anderson JD, Widom J. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol Cell Biol 2001; 21:3830 - 9; http://dx.doi.org/10.1128/MCB.21.11.3830-3839.2001; PMID: 11340174
- Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nat Genet 2006; 38:1210 - 5; http://dx.doi.org/10.1038/ng1878; PMID: 16964265
- Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, Moore IK, et al. A genomic code for nucleosome positioning. Nature 2006; 442:772 - 8; http://dx.doi.org/10.1038/nature04979; PMID: 16862119
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, et al. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 2011; 333:1758 - 60; http://dx.doi.org/10.1126/science.1206097; PMID: 21940898
- Hennig BP, Bendrin K, Zhou Y, Fischer T. Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. EMBO Rep 2012; 13:997 - 1003; http://dx.doi.org/10.1038/embor.2012.146; PMID: 23032292
- Pointner J, Persson J, Prasad P, Norman-Axelsson U, Strålfors A, Khorosjutina O, et al. CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J 2012; 31:4388 - 403; http://dx.doi.org/10.1038/emboj.2012.289; PMID: 23103765
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell 2003; 11:1587 - 98; http://dx.doi.org/10.1016/S1097-2765(03)00231-4; PMID: 12820971
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol Cell 2004; 14:667 - 73; http://dx.doi.org/10.1016/j.molcel.2004.05.013; PMID: 15175161
- Fazzio TG, Tsukiyama T. Chromatin remodeling in vivo: evidence for a nucleosome sliding mechanism. Mol Cell 2003; 12:1333 - 40; http://dx.doi.org/10.1016/S1097-2765(03)00436-2; PMID: 14636590
- Whitehouse I, Tsukiyama T. Antagonistic forces that position nucleosomes in vivo. Nat Struct Mol Biol 2006; 13:633 - 40; http://dx.doi.org/10.1038/nsmb1111; PMID: 16819518
- Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 1997; 90:145 - 55; http://dx.doi.org/10.1016/S0092-8674(00)80321-9; PMID: 9230310
- Jiang C, Pugh BF. A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol 2009; 10:R109; http://dx.doi.org/10.1186/gb-2009-10-10-r109; PMID: 19814794
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 2005; 309:626 - 30; http://dx.doi.org/10.1126/science.1112178; PMID: 15961632
- Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, et al. Nucleosome organization in the Drosophila genome. Nature 2008; 453:358 - 62; http://dx.doi.org/10.1038/nature06929; PMID: 18408708
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 2008; 18:1051 - 63; http://dx.doi.org/10.1101/gr.076463.108; PMID: 18477713
- Pecinka A, Dinh HQ, Baubec T, Rosa M, Lettner N, Mittelsten Scheid O. Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 2010; 22:3118 - 29; http://dx.doi.org/10.1105/tpc.110.078493; PMID: 20876829
- Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, White J, et al. An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 2006; 442:295 - 8; http://dx.doi.org/10.1038/nature04884; PMID: 16855589
- Haag JR, Pikaard CS. Multisubunit RNA polymerases IV and V: purveyors of non-coding RNA for plant gene silencing. Nat Rev Mol Cell Biol 2011; 12:483 - 92; http://dx.doi.org/10.1038/nrm3152; PMID: 21779025
- Jia Y, Lisch DR, Ohtsu K, Scanlon MJ, Nettleton D, Schnable PS. Loss of RNA-dependent RNA polymerase 2 (RDR2) function causes widespread and unexpected changes in the expression of transposons, genes, and 24-nt small RNAs. PLoS Genet 2009; 5:e1000737; http://dx.doi.org/10.1371/journal.pgen.1000737; PMID: 19936292
- Dorweiler JE, Carey CC, Kubo KM, Hollick JB, Kermicle JL, Chandler VL. mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell 2000; 12:2101 - 18; PMID: 11090212
- Haring M, Bader R, Louwers M, Schwabe A, van Driel R, Stam M. The role of DNA methylation, nucleosome occupancy and histone modifications in paramutation. [Epub ahead of print] Plant J 2010; http://dx.doi.org/10.1111/j.1365-313X.2010.04245.x; PMID: 20444233
- Louwers M, Bader R, Haring M, van Driel R, de Laat W, Stam M. Tissue- and expression level-specific chromatin looping at maize b1 epialleles. Plant Cell 2009; 21:832 - 42; http://dx.doi.org/10.1105/tpc.108.064329; PMID: 19336692
- Stam M, Belele C, Dorweiler JE, Chandler VL. Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev 2002; 16:1906 - 18; http://dx.doi.org/10.1101/gad.1006702; PMID: 12154122
- Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS. Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc Natl Acad Sci U S A 2001; 98:9161 - 6; http://dx.doi.org/10.1073/pnas.151244298; PMID: 11470895
- Springer NM, Ying K, Fu Y, Ji T, Yeh CT, Jia Y, et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet 2009; 5:e1000734; http://dx.doi.org/10.1371/journal.pgen.1000734; PMID: 19956538
- Gupta S, Dennis J, Thurman RE, Kingston R, Stamatoyannopoulos JA, Noble WS. Predicting human nucleosome occupancy from primary sequence. PLoS Comput Biol 2008; 4:e1000134; http://dx.doi.org/10.1371/journal.pcbi.1000134; PMID: 18725940
- Arteaga-Vazquez M, Sidorenko L, Rabanal FA, Shrivistava R, Nobuta K, Green PJ, et al. RNA-mediated trans-communication can establish paramutation at the b1 locus in maize. Proc Natl Acad Sci U S A 2010; 107:12986 - 91; http://dx.doi.org/10.1073/pnas.1007972107; PMID: 20616013
- Dalmay T, Hamilton A, Rudd S, Angell S, Baulcombe DC. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 2000; 101:543 - 53; http://dx.doi.org/10.1016/S0092-8674(00)80864-8; PMID: 10850496
- Mourrain P, Béclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, et al. Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell 2000; 101:533 - 42; http://dx.doi.org/10.1016/S0092-8674(00)80863-6; PMID: 10850495
- Deboo GB, Albertsen MC, Taylor LP. Flavanone 3-hydroxylase transcripts and flavonol accumulation are temporally coordinate in maize anthers. Plant J 1995; 7:703 - 13; http://dx.doi.org/10.1046/j.1365-313X.1995.07050703.x; PMID: 7773305
- Goff SA, Klein TM, Roth BA, Fromm ME, Cone KC, Radicella JP, et al. Transactivation of anthocyanin biosynthetic genes following transfer of B regulatory genes into maize tissues. EMBO J 1990; 9:2517 - 22; PMID: 2369901
- Labonne JJ, Goultiaeva A, Shore JS. High-resolution mapping of the S-locus in Turnera leads to the discovery of three genes tightly associated with the S-alleles. Mol Genet Genomics 2009; 281:673 - 85; http://dx.doi.org/10.1007/s00438-009-0439-5; PMID: 19283410
- Steinmüller K, Apel K. A simple and efficient procedure for isolating plant chromatin which is suitable for studies of DNase I-sensitive domains and hypersensitive sites. Plant Mol Biol 1986; 7:87 - 94; http://dx.doi.org/10.1007/BF00040135
- Thuring RW, Sanders JP, Borst P. A freeze-squeeze method for recovering long DNA from agarose gels. Anal Biochem 1975; 66:213 - 20; http://dx.doi.org/10.1016/0003-2697(75)90739-3; PMID: 1096670