Abstract
Genetic and epigenetic alterations in cervical carcinomas were investigated using NotI-microarrays containing 180 cloned sequences flanking all NotI-sites associated with genes on chromosome 3. In total, 48 paired normal/tumor DNA samples, specifically enriched in NotI-sites, were hybridized to NotI-microarrays. Thirty genes, including tumor suppressors or candidates (for example, VHL, RBSP3/CTDSPL, ITGA9, LRRC3B, ALDH1L1, EPHB1) and genes previously unknown as cancer-associated (ABHD5, C3orf77, PRL32, LOC285375, FGD5 and others), showed methylation/deletion in 21–44% of tumors. The genes were more frequently altered in squamous cell carcinomas (SCC) than in adenocarcinomas (ADC, p < 0.01). A set of seven potential markers (LRRN1, PRICKLE2, VHL, BHLHE40, RBSP3, CGGBP1 and SOX14) is promising for discrimination of ADC and SCC. Alterations of more than 20 genes simultaneously were revealed in 23% of SCC. Bisulfite sequencing analysis confirmed methylation as a frequent event in SCC. High down-regulation frequency was shown for RBSP3, ITGA9, VILL, APRG1/C3orf35 and RASSF1 (isoform A) genes (3p21.3 locus) in SCC. Both frequency and extent of RASSF1A and RBSP3 mRNA level decrease were more pronounced in tumors with lymph node metastases compared with non-metastatic ones (p ≤ 0.05). We confirmed by bisulfite sequencing that RASSF1 promoter methylation was a rare event in SCC and, for the first time, demonstrated RASSF1A down-regulation at both the mRNA and protein levels without promoter methylation in tumors of this histological type. Thus, our data revealed novel tumor suppressor candidates located on chromosome 3 and a frequent loss of epigenetic stability of 3p21.3 locus in combination with down-regulation of genes in cervical cancer.
Introduction
Cervical cancer (CC) is the second most common cause of cancer deaths in women worldwide. Molecular as well as epidemiological studies demonstrate that high-risk human papillomaviruses (HPV) are the causative agents for cervical cancer. The recently introduced vaccines can prevent the initial infection by two of these high-risk types, HPV 16 and 18, which are responsible for about 70% of cervical cancers.Citation1 These vaccines provide very effective protection in previously non-exposed women, but they do not seem to possess a significant therapeutic effect in already infected individuals. Not all patients with HPV infection develop invasive lesions; thus, molecular tests for cervical cytology screening may help to identify women with increased risk for progression to cervical carcinoma. Based on this knowledge, a retrieval of new biomarkers for dysplastic cervical cells and additional signaling pathways targeted in malignant progression is necessary.
The important role of chromosome 3 in cancer is well known, its short arm (3p) harbors several regions that include many known tumor suppressor genes (TSGs) and TSG candidates.Citation2,Citation3 Earlier, we performed a comprehensive deletion survey of 3p in more than 400 samples of major epithelial tumors, including CC and identified two most frequently affected regions (“hotspots”) – LUCA at the centromeric and AP20 at the telomeric borders of the 3p21.3 locus.Citation4,Citation5 It was shown that different genetic alterations of this locus are associated with cervical carcinogenesis.
At the present time methylation of CpG islands in promoter regions is known to be precisely regulated during cell differentiation, and plays a key role in the control of gene expression and in cancer (for a review see ref. Citation6). CpG islands are located in promoter regions of many genes associated with cancer and its hypermethylation has been observed as a frequent mechanism of TSGs inactivation, which contributes to malignant transformation. A comprehensive analysis of methylation status of chromosome 3 in CC was not still performed.
Recently, we developed a novel technology based on NotI-microarrays (NMA) and successfully used it for simultaneous analysis of deletions/methylations in lung,Citation7 ovaryCitation8 and colorectal cancer.Citation9 NotI-microarrays can serve as a valued complement of the new techniques for advanced search of methylated cancer-associated genes, first of all TSGs. Moreover, NMA are the unique tools that permit to identify both structural (deletions) and epigenetic (methylation) changes simultaneously. This methodology has been described in detail earlier.Citation8,Citation10 Briefly, the essence of this method consists of the ability of the NotI restriction enzyme to recognize and digest only the unmethylated CG-rich motif GCGGCCGC often found in CpG islands associated with regulatory region of many genes (). The technique involves a special procedure of isolation of genomic DNA fragments flanking by NotI-digested sites from total tumor/normal DNA and usage of them as a NotI-enriched probe (100–500 bp) for comparative hybridization on microarrays containing all 180 NotI-associated clones from a NotI-library of chromosome 3.Citation11,Citation12 Lack or decrease of hybridization signals of tumor DNA compared with normal DNA indicates the deletion or methylation of NotI-associated DNA fragments.
Figure 1. The scheme of DNA hybridization on NotI-microarrays. (A) isolation of genomic DNA. (B) DNA digestion with methyl-specific rare-cutter enzyme NotI. (C) Ligation of the fragments to NotI-linkers containing biotin. (D) DNA digestion with 4-base restriction enzyme Sau3AI. (E) fragments conjugation to microbeads containing streptavidin and washing. (F) ligation of fragments to Sau3AI-linkers containing PCR primer-annealing site. (G) PCR amplification of DNA sequences that has been attached to the microbeads. Then the standard procedures can also be performed: subtraction hybridization in order to remove abundant DNA fragments and to enrich the probe with further sequencing. (H) NotI-enriched probe hybridization to microarrays.
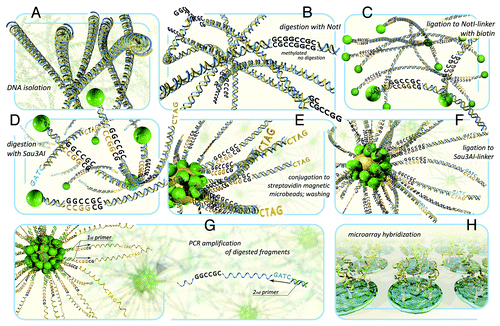
The aim of our study was to identify new genes using NMA technology that can be potential markers for CC. For the first time, 30 genes have been revealed to be methylated/deleted on chromosome 3 in more than 20% of cervical primary tumors. For the majority of these genes, methylation and/or deletions (M/Ds) were not described in CC earlier. The NotI-microarray data were confirmed for a portion of revealed TGSs and candidate genes by bisulfite genomic sequencing and qPCR expression analysis. We also found a novel set of genes with epigenetic alterations that may be useful for discrimination between cervical adenocarcinoma (ADC) and squamous cell carcinoma (SCC) cells. In addition, for the first time we showed the frequent down-regulation of well-known TSG, RASSF1 (isoform A), without RASSF1 promoter methylation that was specific for SCC of cervix. We showed that deletions and epigenetic inactivation of TSGs in chromosome 3 are frequent mechanisms in CC.
Results
Analysis of methylation/deletion frequency using NotI-microarrays
Chromosome 3 specific NMA containing 180 NotI linking clones associated with 188 genes were hybridized with NotI-enriched DNA probes from paired normal/tumor samples ( and ; Table S1). The statistical analysis showed 30 genes with M/D in more than 20% of the cervical cancer samples. The patterns of aberrations were different for two histological types - ADC and SCC. In general, genes were more frequently methylated/deleted in SCC than in ADC samples (p < 0.01) ( and ). We revealed that at least 9 out 39 (23%) tested cervical SCC samples had alterations of more than 20 genes simultaneously including genes from Ap20 and LUCA sub-regions (, samples 11, 17, 20, 25, 27, 38, 42, 47 and 48). Among genes frequently methylated/deleted in CC, only a few were already known TSGs, for example, RBSP3/CTDSPL, ITGA9 and VHL. The majority of found genes were previously not shown to be involved in cervical carcinogenesis, among them LOC285205, FGD5, RPL32, ROPN1, CGGBP1, NBEAL2 and LOC285375 (see Table S2 for information about functions of the corresponding proteins and their association with carcinogenesis).
Figure 2. Hybridization pattern of DNA from cervical cancer samples on NotI-microarrays. (A) Vertically—180 NotI-sites arranged according their localization on chromosome 3 (from 3p26.2 to 3p11.1 and from 3q11.2 to 3q29). Horizontally—48 cervical cancer samples (9 ADC and 39 SCC). (B) Vertically—41 NotI-sites arranged by methylation/deletion frequency (from 44% to 15%). Numbers correspond to numbers from . *Samples from patients with lymph node metastases.
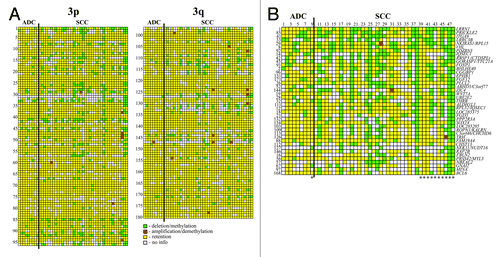
Table 1. List of chromosome 3 NotI-sites with methylation/deletion frequencies more than 20% in cervical cancer
Confirmation of NotI-microarrays results by bisulfite genomic sequencing
To confirm the results of NMA hybridizations, methylation status of seven genes with frequency of M/D 27–44% according NMA were analyzed in 11 SCC samples by bisulfite sequencing (namely LRRN1, ITGA9, LRRC3B, NKIRAS1, VHL, RBSP3 and FGF12). The results of bisulfite sequencing are represented in and Tables S3–S5. Methylation of CpG promoter islands was confirmed in most of tested cases (19 out of 23). For 4 cases (LRRN1 Nº42, NKIRAS1 Nº17, VHL Nº15 and RBSP3 Nº17), the reason of decreased hybridization signal at NotI-microarray was another than NotI-site methylation (number of samples in parentheses as ). According to our previous qPCR data, deletions are the main mechanism of NKIRAS1 gene inactivation.Citation13 Probably, hemizygous deletions took place for three other unmethylated cases although a loss of NotI-sites due to point mutations cannot be excluded. So, among 23 randomly selected cases from NotI-panel with decreased signal of hybridization 19 cases (83%) showed methylation of gene CpG islands including NotI recognition sites. Thus, bisulfite sequence data are in good concordance with NotI-microarrays results and suggest that methylation of 5′ regulator regions of genes is a frequent event in SCC.
Table 2. Analysis of methylation status of 7 genes in cervical carcinomas from NMA panel by bisulfite genomic sequencing
Cervical SCC and ADС have different methylation/deletion patterns
We observed the difference of M/D patterns in SCC and ADC for 22 tested genes (). However, only for two genes, PPP2R3A and LRRN1, this difference was statistically valid (p = 0.03 for both). But this difference was obvious for all 22 genes taken together (): mean frequency of methylation/deletion decreased from 37% for SCC to 5% for ADC samples (p < 0.01). The statistical analysis of our data suggested the most promising set of 7 genes (LRRN1, PRICKLE2, VHL, BHLHE40, RBSP3, CGGBP1 and SOX14) that discriminate SCC and ADC. The revealing of alteration in two and more of these genes in a sample would indicate the presence of SCC cells but not ADC. The sensitivity of the set is equal to (67 ± 8) % and the specificity is 100%.
Simultaneous down-regulation of RBSP3, ITGA9, VILL, APRG1/C3orf35 and RASSF1A genes in cervical SCC
Earlier, we demonstrated that expression loss of several genes from both LUCA and AP20 sub-regions within the 3p21.3 locus occurred frequently and simultaneously in the same tumor in lung cancer (p < < 0.01).Citation14 Taking into account these data and a frequent simultaneous genetic/epigenetic destabilization of many genes at the 3p () we expected that analysis of NotI-sites will reveal a wide region of epigenetic destabilization where neighboring genes without NotI-sites in their CpG islands might also be down-regulated. To evaluate the rate of down-regulation of genes that may be simultaneously destabilized we have quantified and compared mRNA levels for several genes from 3p21.3 locus in SCC samples. We included in this analysis four genes from AP20 sub-region: two NotI–associated TSGs, RBSP3 and ITGA9, and two candidate genes, VILL and APRG1, without NotI-sites in their regulatory regions. RBSP3 and ITGA9 are known to be involved in carcinogenesis and frequently methylated/deleted in many cancers, including cervical SCC according our NotI-microarray data (). Though genes VILL and APRG1 are localized in AP20, the data confirming their involvement in carcinogenesis are rather poor. We analyzed also well-known TSG RASSF1A from the LUCA sub-region. RASSF1 has no NotI-sites in the promoter region, but we have selected it taking into account the following points: 1) its inactivation caused by promoter methylation has been demonstrated in many types of tumors; 2) published data about the RASSF1 frequency of methylation in СС are contradictory (for a review seeCitation15).
All five genes were down-regulated (2–78-fold) in the majority of cases of SCC (53–89%, and ). High frequencies of aberrations were inherent both for three known TSGs from two “hotspots” and too poorly studied genes. High frequency (60%) and extent of the APRG1 mRNA decrease (up to 18-fold) indicates on the need for further research of this gene ().
Figure 3. The relative expression level (R) of five genes (3р21.3) in SCC with different clinical characteristics. Results of qPCR. Samples without lymph node metastases (stages I and II), samples with lymph node metastases (stage III). *APRG1 mRNA was not detected in both tumor and adjacent normal tissues.
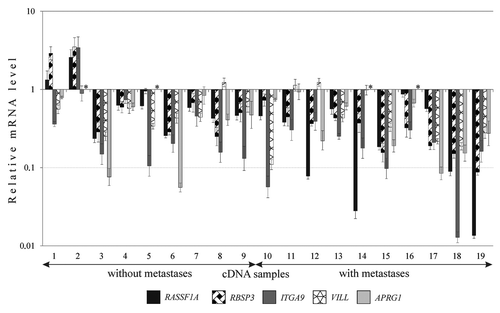
Table 3. QPCR analysis of five genes (3p21.3) in cervical SCC
Simultaneous decrease of mRNA levels of tested genes was observed in all SCC samples, except two ones (samples 1 and 2, ). Moreover, four genes (except, APRG1) revealed the tendency to increase frequency and extent of mRNA decrease in samples from patients with lymph nodes metastases compared with samples without metastases (). For RASSF1A and RBSP3, this difference was statistically significant (p ≤ 0.05). Three TSGs, RASSF1A, RBSP3 and ITGA9, showed concordant expression profiles: we observed a positive correlation between their mRNA levels. Spearmen rank coefficient values reflecting the correlation between mRNA levels were: 0.69 for RASSF1A and RBSP3 (p < 0.01), 0.43 for RBSP3 and ITGA9 (p = 0.07), and 0.48 for RASSF1A and ITGA9 (p = 0.04).
The frequencies of mRNA level decrease and NotI methylation/deletions were compared in the same samples for two genes containing NotI-sites, namely, RBSP3 and ITGA9. The RBSP3 mRNA level was decreased in 68% of SCC samples and methylation/deletions were detected in 36%; for the ITGA9 gene these values were 89% and 44%, respectively. Thus, these findings were in concordance with the observed frequent inactivation of the genes highlighted by NotI-microarray and suggest the existence of additional mechanisms of their inactivation besides of DNA methylation.
RASSF1A down-regulation of is not associated with hypermethylation of its promoter in cervical SCC
We revealed the decrease of the RASSF1A mRNA level in 58% (11 out 19) of cervical SCC ( and ). It was an unexpected result because according several reports RASSF1 promoter methylation was rare event in SCC of cervix in opposition to ADC (for a review seeCitation15). To understand the reasons of RASSF1A down-regulation, we estimated its mRNA level by qPCR and the methylation status of the RASSF1 promoter by bisulfite sequencing in 17 primary SCC cervical tumors and 4 cervical carcinoma cell lines ( and ). Surprisingly the promoter methylation was detected only in the HPV-negative C33A cell line, that was used as positive control.Citation16 All primary tumors with the RASSF1A mRNA level decrease and 3 HPV-positive SCC cell lines demonstrated sporadic methylation of a few CpG sites of the RASSF1 promoter. We did not revealed substantial methylation of the RASSF1 promoter even in tumors with nearly complete absence of the RASSF1A mRNA (samples 16 and 17, ). There is no difference in methylation patterns between healthy cervical tissue, SCC cell lines with more than 2-fold decrease of mRNA levels (C4–1, CaSki) and SiHa cells with unchanged mRNA level compared with healthy cervix ( and ).
Figure 4. Analysis of methylation status of RASSF1 promoter in cervical SCC samples and cell lines. Schematic representation of results of sodium bisulfite sequencing: numbers under the horizontal line indicate CpG dinucleotides within RASSF1 promoter, the broken arrow – transcription initiation site; black circle,methylated CpG; white circle, unmethylated CpG; gray circle, partially methylated CpG; each row of circles represents a cloned DNA molecule for all samples excepting SiHa and number 16T, where the total PCR products are presented; T, tumor; Nc, normal cervix. Numbers of tumors correspond to .

Table 4.RASSF1 methylation status and relative mRNA levels of RASSF1A in cervical SCC and cell lines
Figure 5. Analysis of RASSF1A protein and mRNA levels in cervical cancer cell lines. (A) Left column: immunochemical detection of RASSF1А; right column – the same as left column plus DAPI staining of nuclei (blue). (B) Results of qPCR. Columns mean averaged RASSF1A mRNA level of three different propagations of each cell line. All calculations were performed relatively to average value of three HPV-negative healthy cervical tissues.
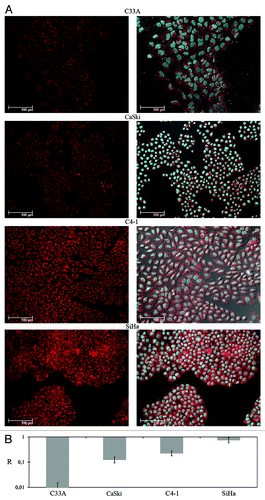
Then the RASSF1A protein content was estimated in 4 cell lines with different RASSF1A mRNA levels by immunohistochemistry. Three cell lines demonstrated the decrease of cytoplasmic staining intensity compared with SiHa cells with unchanged mRNA level ( and ). The extent of fluorescence signal was correlated with the RASSF1A mRNA level. The strongest protein down-regulation was detected for cell line C33A, in which RASSF1 promoter was methylated, but substantial decrease of protein levels were detected in cell lines without promoter methylation (CaSki, C4–1).
Thus using bisulfite sequencing we have confirmed that RASSF1 promoter methylation is a rare event in cervical SCC and for the first time have demonstrated its down-regulation at both mRNA and protein levels without promoter methylation in tumors of this histological type. In addition these findings indicate that another mechanisms besides DNA methylation may be destabilized in the region of the RASSF1 gene as well as in regions of RBSP3 and ITGA9 genes.
Discussion
In this study, using original technology of NotI-microarrays,Citation10 we found 30 genes on chromosome 3, including known TSGs and TSG-candidates (for example, VHL, RBSP3, ITGA9, LRRC3B, ALDH1L1, EPHB1) and also genes which have not been previously reported as involved in cancer development (ABHD5, C3orf77, PRL32, LOC285375, FGD5, etc.) with high frequencies (more than 20%) of methylation/deletion during cervical carcinogenesis ( and S1). Many of these genes (LOC285205, FGD5, RPL32, ROPN1, CGGBP1, NBEAL2, LOC285375 and others) were not shown to be involved in cervical carcinogenesis previously, but according to our recent data many of them were involved in the development of non-small cell lung and ovarian cancer.Citation7,Citation8 These findings suggest that genetic and epigenetic destabilization of genes at chromosome 3 is common mechanisms of epithelial tumors development. According literature data, the protein products of newly identified genes belong to pathways affected during development and progression of different cancer types (Table S2): in WNT signaling pathway (E3 ubiquitin-protein ligase, PDZRN3), in the regulation of cellular polarity and invasion (PRICKLE2), in regulation of the actin cytoskeleton (FGD5), in MAP kinase signaling pathway (FGF12) and others. Some of them are transcription factors involved in regulation of apoptosis (BHLHE40) and tissue-specific expression (FOXP1). Functions of some proteins are currently unknown, for example, LRRN1, LRRC3B and C3orf77. High frequencies of methylation/deletions, participation in signaling pathways affected in many tumors and inactivation at least in three types of epithelial tumors according our data argue in favor of tumor suppression functions of these genes.
Two out of 30 genes (PDZRN3 and SOX14) had aberrations only in cervical cancer, but not lung and ovarian. PDZRN3 plays an important role in regulation of the surface level of MUSK (skeletal muscle-specific tyrosine-protein kinase receptor) on myotubules. SOX14 is involved in the regulation of embryonic development and in the determination of the cell fate (Table S2). These genes might be potentially used for discrimination between different cancer types.
We detected that the patterns of aberrations were different for two histological types of CC—ADC and SCC. In general, squamous cell tumors revealed higher rates of methylation/deletions than adenocarcinomas (p < 0.01). It is known that, in spite of general decline of CC incidence, the contribution of cervical ADC, absolute and relative to SCC, has been rising steadily in many countries, especially for younger women. This may be a result of the limitations of ADC detection during СС screening: adenocarcinomas are often diagnosed at an advanced disease stage (for a review see ref. Citation17). The statistical analysis of our data suggested the most predictive set of seven genes (LRRN1, PRICKLE2, VHL, BHLHE40, RBSP3, CGGBP1 and SOX14) that may discriminate SCC and ADC with accuracy (73 ± 6) %. These genes might be useful for analysis by PCR-based technique on early stages of disease.
We have validated our microarray data for genes LRRN1, ITGA9, LRRC3B, NKIRAS1, VHL, RBSP3 and FGF12 by bisulfite genomic sequencing and have revealed that methylation of tested genes takes place in CC and is a prevalent event (83% of tested cases) compared with deletions that are also detected by NotI-microarrays. These results, and our recent data obtained for lung and ovarian cancers using NotI-microarrays,Citation7,Citation8 suggest that methylation of promoter regions is the important mechanism leading to inactivation of TSG and TSG-candidates on chromosome 3.
We found out a group of cervical SCC (23% of cases) in which more than 20 NotI-sites were affected simultaneously (). The majority of the corresponding genes is located on 3p, the short arm of chromosome 3. It is known that groups of tumors with high degrees of methylation (the CpG island methylator phenotype, or CIMP+) exist in colorectal and some other types of cancers.Citation18 CIMP+ tumors represent a clinically distinct group of cancers that is characterized by epigenetic instability. Our data suggest that CIMP+ and epigenetic instability on 3p are characteristic features of a portion of HPV-positive cervical SCC. Earlier hypermethylation of some genes have been identified not only in cervical cancer but also in its precursors, suggesting that assays for these molecular events might be clinically useful for cytological screening and diagnostics of CC at early stages.Citation19-Citation22 Therefore, the usage of methylation markers (known and newly identified) in addition to conventional cytological screening and human papillomavirus testing may help to reveal cancer cells with CIMP+.
To confirm epigenetic deregulation of 3p21.3 locus, we evaluated mRNA levels of five genes from this sub-region, RBSP3, ITGA9, APRG1, VILL and RASSF1A, in cervical SCC. High frequencies (53–89%) of down-regulation of all these genes were revealed for the first time in cervical SCC excepting RBSP3.Citation23,Citation24 Along with known TSGs, RASSF1A, RBSP3 and ITGA9 (see Table S2), two poorly studied genes, APRG1 and VILL, were strongly down-regulated in 60% and 53% of cervical tumors respectively. There is only scarce information about their functions and involvement in cancer. APRG1 is located proximal to the border of the homozygous deletion that takes place in a small cell lung cancer cell line ACC-LC5.Citation25 APRG1 was suggested to have tumor suppression function in breast cancer.Citation26 This gene encodes a putative protein of 170 amino acids (isoform B), which has an N-terminal part conserved among members of the eukaryotic translation factor 6 gene family. The analysis by PROSITE, the database of protein domains, families and functional sites, had revealed peptide patterns corresponding to the cell attachment sequence Arg-Gly-Asp, which suggests the involvement of APRG1 in membrane interactions and cell adhesion.Citation25 Тhe protein encoded by VILL belongs to the villin/gelsolin family. It is required for actin bundle assembly.Citation27 No association between VILL and tumorigenesis has been revealed, but villin 1, a member of this family, was reported to be either up- or down-regulated in different kinds of cancer.Citation28,Citation29 Our data suggest that these genes could be considered as TSGs candidates. Further research of their functions in carcinogenesis is necessary.
It was surprising that we detected inactivation of RASSF1A in 58% of cervical SCC. Previous published data concerning to RASSF1 methylation status in CC were contradictory. A number of publications demonstrated that methylation of its promoter took place in cervical ADC but was a very rare event in cervical SCC. On the other hand, frequent RASSF1 promoter methylation in cervical SCC and precancerous lesions was described in several publications (for a review seeCitation15). Unfortunately, levels of RASSF1A expression in combination with RASSF1 promoter methylation were not evaluated in these studies. In our experiments, we did not revealed pronounced promoter methylation in any tumor out of 10 SCC with decreased mRNA levels (). The RASSF1A inactivation at the mRNA and protein levels correlated with RASSF1 promoter methylation only in one out of 4 cervical SCC cell lines (; and 4). Thus, we have confirmed that the RASSF1 promoter methylation is a rare event in cervical SCC and, for the first time, have demonstrated frequent RASSF1A down-regulation without RASSF1 promoter methylation in tumors of this histological type.
Well-known TSGs, RASSF1A, RBSP3 and ITGA9, showed coordinated down-regulation in the same SCC patients. Earlier, we demonstrated simultaneous inactivation of some genes from this 3p21.3 region, including RASSF1A, RBSP3 and ITGA9 in lung cancer.Citation14,Citation30 These results point to the importance of this region for epithelium tumor development and allow to assume the existence of common mechanisms of gene down-regulation in this locus. Several groups of data indicate the existence of other epigenetic mechanisms, besides DNA methylation, inactivating these genes. We clearly demonstrated that at least one gene from the 3p21.3 locus, RASSF1A, was frequently silenced in the absence of promoter methylation in cervical SCC. We confirmed, using bisulfite genomic sequencing, that promoter methylation of RBSP3 and ITGA9 took place in a portion of CC; on the other hand, we demonstrated that the frequency of down-regulation revealed by qPCR was significantly higher than methylation/deletions rate showed by NotI-microarray (68 and 31% for RBSP3; 89 and 42% for ITGA9, respectively). The same results were obtained for these genes in lung cancer: the frequency of RBSP3 and ITGA9 mRNA level decreases (85%, for each gene) was also higher than the frequency of methylation/deletions (47.5 and 45%, respectively).Citation7 The lack of concordance between these results can be related to participation of chromatin modification and RNA interference in gene inactivation instead of, or in addition to, DNA methylation. To predict microRNA regulation of RASSF1, RBSP3 and ITGA9 genes, we used miRNA body map (www.mirnabodymap.org/index.php),Citation31 which includes data from five microRNA resources (TargetScan, DIANA, PITA, miRDB and MicroCosm) implementing different algorithms of microRNA target prediction. The analysis demonstrated that each of the three mRNAs (RASSF1, RBSP3 and ITGA9) may be the target of three microRNAs (miR-346, miR-515 and miR-767–5p) simultaneously. All these miRNAs seem to play a role in tumors onset or progression. The miR-346 is overexpressed in follicular thyroid carcinomaCitation32 and in serum of prostate cancer patients.Citation33 In vitro overexpression of miR-346 induced proliferation, whereas inhibition of its expression led to growth arrest.Citation32 miR-515, a precursor of miR-515–5p, was overexpressed in oral carcinomas.Citation34 The third microRNA, miR-767–5p, was found to play a major role in oncogenic processes in different tumors. Deregulation of any of these miRNAs might result in coordinate inactivation of the three genes. Thus, the relations of these miRNAs to the regulation of RASSF1, RBSP3 and ITGA9 need to be studied. Recently, it was demonstrated that epigenetic silencing can span large regions of the chromosome in prostate and colorectal cancer. Both methylated DNA and neighboring unmethylated genes can be coordinately suppressed by global changes in histone modification across an entire chromosome band within chromosomes 2 and 7 in these tumors.Citation35,Citation36 These large regions were associated with regional histone deacetylation combined with subdomains of different epigenetic patterns, which include re-enforcement, gain or exchange of repressive histone, and DNA methylation marks. Authors believe that coordinate epigenetic control over larger regions may be a common phenomenon in cells during development and may be involved in cancer. Our data suggest that the 3p21.3 locus might be such long-range epigenetic silencing region in CC () and study of its chromatin structure in combination with DNA methylation should be interesting.
In general, our study revealed a frequent loss of epigenetic stability in combination with gene down-regulation in 3p21.3 locus in CC. This study reasserts that NotI-microarrays are powerful tools for disclosing TSG candidates and provides a basis for better understanding of mechanisms involved in development of CC.
Materials and Methods
Tissue specimens and cell lines
Paired specimens of tissues including 39 squamous cell carcinomas (SCC), 9 adenocarcinomas (ADC) of uterine cervix and adjacent morphologically normal tissues (conventional “normal” tissues) were obtained after surgical resection prior radiation or chemotherapy and stored in liquid nitrogen. An additional set of SCC (19 paired normal/tumor tissues) was used for further validation by qPCR. Tissues were obtained from patients with FIGO stages I, II and III of the disease at the N.N. Blokhin Cancer Research Center. The diagnosis was verified by histopathology and only samples containing 70–80% or more tumor cells were used in the study. All tumors were E7 HPV16/18-positive according to PCR analysis. Sample information is represented in . Tissues of healthy cervix were obtained from patients with uterine polyps or tumors and tested for HPV absence. All tissues were collected under the approval of the Institutional Review Board of the Blokhin Cancer Research Center. Informed consent was obtained from all patients. The study was done in accordance with the principles outlined in the Declaration of Helsinki. The HPV-positive human cervical SCC cell lines SiHa, CaSki, C4–1 and HPV-negative C-33A cell line (American Type Culture Collection) were maintained in DMEM supplemented with 10% FCS.
Table 5. Clinical and pathological characteristics of cervical cancer samples
NotI-microarrays
One hundred eighty NotI linking clones from human chromosome 3 containing 188 genes with inserts up to 15 kb were immobilized on the glass slides in six replications.Citation10,Citation12 Plasmid DNA for immobilization on the glasses was isolated with a HiPure Plasmid Midiprep kit (Invitrogen) and printed on the silanized glasses at a concentration of 0.25 μg/ml with a QarrayMini microarrayer (Genetix, United Kingdom). DNA from E. coli was used as negative hybridization control. Preparation of NotI representations (NotI probes, NR) was done essentially as described previously.Citation37 Hybridization of NR was performed at 42°C for 15 h in a Lucidea Base device (Amersham Pharmacia Biotech) according to manufacturer’s recommendations. Microarrays were scanned in a GenePix 4000A. The technique determines NotI-site methylation or deletion simultaneously. The results were processed with GenePix Pro 6.0 software (Amersham Pharmacia Biotech). Then data were analyzed using our program NIMAN, NotI-Microarray Analysis.Citation7
Bisulfite genomic sequencing
The bisulfite conversion of DNAs was performed using an EZ DNA Methylation Kit (Zymo Research) according to the manufacturer’s instructions. Primers for PCR are available upon request. After amplification of bisulfite treated DNAs PCR products were cloned and used for automated sequencing or were sequenced directly (ABI Prism 3100-Avant Genetic Analyzer, Applied Biosystems).
Quantitative PCR
Total RNA extraction and reverse transcription reaction was done as described earlier.Citation14 The sequences of primers and probes for ITGA9, RBSP3 and RASSF1A genes were published previously.Citation14 For VILL gene we used the following sequences: forward primer – GCACTGACAGCCACAACACCA, reverse primer – ATCACCATTACAGCCCTTCCCA, probe – CCGTGCCTCATCCCTCAACTCCAG; and for APRG1 gene: TTTGGACCCAAGGTAAGAAAACTG, CCATCCAATGCTGTGATTCCAC and ACCAGCCTTCCATTGCTCCACACA respectively. Reference genes GAPDH and RPN1Citation38 were used. All reactions were performed using 7500 Real-Time PCR System (Applied Biosystems). QPCR data were analyzed using the relative quantification or ΔΔCt-method as described.Citation39 Each reaction was repeated three times. At least 2-fold mRNA changes were considered as significant because of reference genes variability.
Immunohistochemistry
RASSF1A protein content in cervical cancer cell lines were estimated with immunohistochemical methodology using confocal microscope (SPES, Leica) with AOTF calibration before each measurement. We used specific primary monoclonal mice antibodies to RASSF1A (Acris) and secondary goat antibodies to mice conjugated with Atto 647 dye, which we chose because of their high photo stability. Fluorescence level in 3D images (reconstructions) was measured using Imaris software.
Statistical analysis
Nonparametric Wilcoxon test was used to compare mRNA expression differences of target and reference genes in cervical cancer samples. Kruskal-Wallis and Mann-Whitney rank-sum tests, Fisher’s exact test and χ2 criteria were used for analysis of genomic DNA copy, methylation and mRNA level changes in cervical cancer groups with different histological characteristics. P-values < 0.05 were considered statistically significant. Spearmen’s rank correlation coefficient was used for revealing correlations between mRNA level changes of different genes. All statistical procedures were performed using our NIMAN software created earlierCitation7 and BioStat software.Citation40 Sensitivity, specificity and accuracy were calculated according to.Citation41 Selection of groups of the genes, allowing potentially to distinguish two main histological types of CC - SCC and ADC was performed by often used statistical method SVM (support vector machine). The number of genes was chosen to create a set with the maximum distance between these two groups.
Additional material
Download Zip (441.2 KB)Acknowledgments
This work was supported by State Contract 16.552.11.7069 with the Russian Ministry of Education and Science, Grants 07-04-00708 and 13-04-01885 from the Russian Foundation for Basic Research, The National Academy of Sciences of Ukraine, Ukranian State Foundation of Fundamental Research F46/457–2011, and State Agency on Science, Innovations and Informatization of Ukraine DP/487–2012.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- zur Hausen H. Papillomaviruses--to vaccination and beyond. Biochemistry (Mosc) 2008; 73:498 - 503; http://dx.doi.org/10.1134/S0006297908050027; PMID: 18605974
- Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 2007; 26:7283 - 301; http://dx.doi.org/10.1038/sj.onc.1210547; PMID: 17533367
- Ji L, Minna JD, Roth JA. 3p21.3 tumor suppressor cluster: prospects for translational applications. Future Oncol 2005; 1:79 - 92; http://dx.doi.org/10.1517/14796694.1.1.79; PMID: 16555978
- Senchenko V, Liu J, Braga E, Mazurenko N, Loginov W, Seryogin Y, et al. Deletion mapping using quantitative real-time PCR identifies two distinct 3p21.3 regions affected in most cervical carcinomas. Oncogene 2003; 22:2984 - 92; http://dx.doi.org/10.1038/sj.onc.1206429; PMID: 12771950
- Senchenko VN, Liu J, Loginov W, Bazov I, Angeloni D, Seryogin Y, et al. Discovery of frequent homozygous deletions in chromosome 3p21.3 LUCA and AP20 regions in renal, lung and breast carcinomas. Oncogene 2004; 23:5719 - 28; http://dx.doi.org/10.1038/sj.onc.1207760; PMID: 15208675
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27 - 36; http://dx.doi.org/10.1093/carcin/bgp220; PMID: 19752007
- Dmitriev AA, Kashuba VI, Haraldson K, Senchenko VN, Pavlova TV, Kudryavtseva AV, et al. Genetic and epigenetic analysis of non-small cell lung cancer with NotI-microarrays. Epigenetics 2012; 7:502 - 13; http://dx.doi.org/10.4161/epi.19801; PMID: 22491060
- Kashuba V, Dmitriev AA, Krasnov GS, Pavlova T, Ignatjev I, Gordiyuk VV, et al. NotI Microarrays: Novel Epigenetic Markers for Early Detection and Prognosis of High Grade Serous Ovarian Cancer. Int J Mol Sci 2012; 13:13352 - 77; http://dx.doi.org/10.3390/ijms131013352; PMID: 23202957
- Gerashchenko GV, Gordiyuk VV, Skrypkina IY, Kvasha SM, Kolesnik OO, Ugryn DD, et al. Screening of epigenetic and genetic disturbances of human chromosome 3 genes in colorectal cancer. Ukr Biokhim Zh 2009; 81:81 - 7; PMID: 20387637
- Li J, Protopopov A, Wang F, Senchenko V, Petushkov V, Vorontsova O, et al. NotI subtraction and NotI-specific microarrays to detect copy number and methylation changes in whole genomes. Proc Natl Acad Sci U S A 2002; 99:10724 - 9; http://dx.doi.org/10.1073/pnas.132271699; PMID: 12149436
- Zabarovsky ER, Boldog F, Thompson T, Scanlon D, Winberg G, Marcsek Z, et al. Construction of a human chromosome 3 specific NotI linking library using a novel cloning procedure. Nucleic Acids Res 1990; 18:6319 - 24; http://dx.doi.org/10.1093/nar/18.21.6319; PMID: 2243778
- Kashuba VI, Gizatullin RZ, Protopopov AI, Li J, Vorobieva NV, Fedorova L, et al. Analysis of NotI linking clones isolated from human chromosome 3 specific libraries. Gene 1999; 239:259 - 71; http://dx.doi.org/10.1016/S0378-1119(99)00411-4; PMID: 10548727
- Gerashchenko GV, Bogatyrova OO, Rudenko EE, Kondratov AG, Gordiyuk VV, Zgonnyk YM, et al. Genetic and epigenetic changes of NKIRAS1 gene in human renal cell carcinomas. Exp Oncol 2010; 32:71 - 5; PMID: 20693965
- Senchenko VN, Anedchenko EA, Kondratieva TT, Krasnov GS, Dmitriev AA, Zabarovska VI, et al. Simultaneous down-regulation of tumor suppressor genes RBSP3/CTDSPL, NPRL2/G21 and RASSF1A in primary non-small cell lung cancer. BMC Cancer 2010; 10:75; http://dx.doi.org/10.1186/1471-2407-10-75; PMID: 20193080
- Dammann R, Schagdarsurengin U, Seidel C, Strunnikova M, Rastetter M, Baier K, et al. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol 2005; 20:645 - 63; PMID: 15736067
- Kuzmin I, Liu L, Dammann R, Geil L, Stanbridge EJ, Wilczynski SP, et al. Inactivation of RAS association domain family 1A gene in cervical carcinomas and the role of human papillomavirus infection. Cancer Res 2003; 63:1888 - 93; PMID: 12702579
- Seoud M, Tjalma WA, Ronsse V. Cervical adenocarcinoma: moving towards better prevention. Vaccine 2011; 29:9148 - 58; http://dx.doi.org/10.1016/j.vaccine.2011.09.115; PMID: 21983356
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96:8681 - 6; http://dx.doi.org/10.1073/pnas.96.15.8681; PMID: 10411935
- Lendvai A, Johannes F, Grimm C, Eijsink JJ, Wardenaar R, Volders HH, et al. Genome-wide methylation profiling identifies hypermethylated biomarkers in high-grade cervical intraepithelial neoplasia. Epigenetics 2012; 7:1268 - 78; http://dx.doi.org/10.4161/epi.22301; PMID: 23018867
- Overmeer RM, Louwers JA, Meijer CJ, van Kemenade FJ, Hesselink AT, Daalmeijer NF, et al. Combined CADM1 and MAL promoter methylation analysis to detect (pre-)malignant cervical lesions in high-risk HPV-positive women. Int J Cancer 2011; 129:2218 - 25; http://dx.doi.org/10.1002/ijc.25890; PMID: 21190187
- Reesink-Peters N, Wisman GB, Jéronimo C, Tokumaru CY, Cohen Y, Dong SM, et al. Detecting cervical cancer by quantitative promoter hypermethylation assay on cervical scrapings: a feasibility study. Mol Cancer Res 2004; 2:289 - 95; PMID: 15192122
- Widschwendter A, Gattringer C, Ivarsson L, Fiegl H, Schneitter A, Ramoni A, et al. Analysis of aberrant DNA methylation and human papillomavirus DNA in cervicovaginal specimens to detect invasive cervical cancer and its precursors. Clin Cancer Res 2004; 10:3396 - 400; http://dx.doi.org/10.1158/1078-0432.CCR-03-0143; PMID: 15161694
- Anedchenko EA, Kiseleva NP, Dmitriev AA, Kiselev FL, Zabarovskiĭ ER, Senchenko VN. [Tumor suppressor gene RBSP3 in cervical carcinoma: copy number and transcriptional level]. Mol Biol (Mosk) 2007; 41:86 - 95; http://dx.doi.org/10.1134/S0026893307010116; PMID: 17380895
- Mitra S, Mazumder Indra D, Bhattacharya N, Singh RK, Basu PS, Mondal RK, et al. RBSP3 is frequently altered in premalignant cervical lesions: clinical and prognostic significance. Genes Chromosomes Cancer 2010; 49:155 - 70; PMID: 19885927
- Protopopov A, Kashuba V, Zabarovska VI, Muravenko OV, Lerman MI, Klein G, et al. An integrated physical and gene map of the 3.5-Mb chromosome 3p21.3 (AP20) region implicated in major human epithelial malignancies. Cancer Res 2003; 63:404 - 12; PMID: 12543795
- Leris AC, Roberts TR, Jiang WG, Newbold RF, Mokbel K. Evidence for a tumour suppressive function of APRG1 in breast cancer. Breast Cancer Res Treat 2005; 93:97 - 100; http://dx.doi.org/10.1007/s10549-005-4169-z; PMID: 16187228
- Mahajan-Miklos S, Cooley L. The villin-like protein encoded by the Drosophila quail gene is required for actin bundle assembly during oogenesis. Cell 1994; 78:291 - 301; http://dx.doi.org/10.1016/0092-8674(94)90298-4; PMID: 8044841
- Zhang MQ, Lin F, Hui P, Chen ZM, Ritter JH, Wang HL. Expression of mucins, SIMA, villin, and CDX2 in small-intestinal adenocarcinoma. Am J Clin Pathol 2007; 128:808 - 16; http://dx.doi.org/10.1309/JAF3KVGJHQCJ1QF9; PMID: 17951204
- Kennedy MT, Jordan RC, Berean KW, Perez-Ordoñez B. Expression pattern of CK7, CK20, CDX-2, and villin in intestinal-type sinonasal adenocarcinoma. J Clin Pathol 2004; 57:932 - 7; http://dx.doi.org/10.1136/jcp.2004.016964; PMID: 15333652
- Anedchenko EA, Dmitriev AA, Krasnov GS, Kondrat’eva TT, Kopantsev EP, Vinogradova TV, et al. [Down-regulation of RBSP3/CTDSPL, NPRL2/G21, RASSF1A, ITGA9, HYAL1 and HYAL2 genes in non-small cell lung cancer]. Mol Biol (Mosk) 2008; 42:965 - 76; http://dx.doi.org/10.1134/S0026893308060058; PMID: 19140316
- Tsang JS, Ebert MS, van Oudenaarden A. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol Cell 2010; 38:140 - 53; http://dx.doi.org/10.1016/j.molcel.2010.03.007; PMID: 20385095
- Weber F, Teresi RE, Broelsch CE, Frilling A, Eng C. A limited set of human MicroRNA is deregulated in follicular thyroid carcinoma. J Clin Endocrinol Metab 2006; 91:3584 - 91; http://dx.doi.org/10.1210/jc.2006-0693; PMID: 16822819
- Selth LA, Townley S, Gillis JL, Ochnik AM, Murti K, Macfarlane RJ, et al. Discovery of circulating microRNAs associated with human prostate cancer using a mouse model of disease. Int J Cancer 2012; 131:652 - 61; http://dx.doi.org/10.1002/ijc.26405; PMID: 22052531
- Scapoli L, Palmieri A, Lo Muzio L, Pezzetti F, Rubini C, Girardi A, et al. MicroRNA expression profiling of oral carcinoma identifies new markers of tumor progression. Int J Immunopathol Pharmacol 2010; 23:1229 - 34; PMID: 21244772
- Frigola J, Song J, Stirzaker C, Hinshelwood RA, Peinado MA, Clark SJ. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across an entire chromosome band. Nat Genet 2006; 38:540 - 9; http://dx.doi.org/10.1038/ng1781; PMID: 16642018
- Coolen MW, Stirzaker C, Song JZ, Statham AL, Kassir Z, Moreno CS, et al. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat Cell Biol 2010; 12:235 - 46; PMID: 20173741
- Dunwell TL, Hesson LB, Pavlova T, Zabarovska V, Kashuba V, Catchpoole D, et al. Epigenetic analysis of childhood acute lymphoblastic leukemia. Epigenetics 2009; 4:185 - 93; http://dx.doi.org/10.4161/epi.4.3.8752; PMID: 19430199
- Krasnov GS, Oparina NIu, Dmitriev AA, Kudriavtsev AV, Anedchenko EA, Kondrat’eva TT, et al. [Novel reference gene RPN1 for normalization of quantitative data in lung and kidney cancer]. Mol Biol (Mosk) 2011; 45:238 - 48; http://dx.doi.org/10.1134/S0026893311020129; PMID: 21634111
- Senchenko VN, Krasnov GS, Dmitriev AA, Kudryavtseva AV, Anedchenko EA, Braga EA, et al. Differential expression of CHL1 gene during development of major human cancers. PLoS One 2011; 6:e15612; http://dx.doi.org/10.1371/journal.pone.0015612; PMID: 21408220
- Glantz SA. Primer of biostatistics. New York, USA: McGRAW-HILL, 2005.
- Altman DG, Bland JM. Diagnostic tests. 1: Sensitivity and specificity. BMJ 1994; 308:1552; http://dx.doi.org/10.1136/bmj.308.6943.1552; PMID: 8019315